 2015-05-26
2015-05-26 3537
3537Свойства конденсированных систем, обладающих ионной проводимостью. К таким системам относятся: водные растворы электролитов; растворы на основе неводных и так называемых «апротонных» растворителей; обладающих необычными свойствами растворы свободных электронов в жидких средах; растворы полиэлектролитов; расплавы электролитов – ионные жидкости; твердые электролиты, в том числе удивительные твердые электролиты со сверхвысокой проводимостью – суперионики; суперкритические жидкости. Задача теории состоит в том, чтобы количественно описать свойства всех этих систем, как в состоянии равновесия, так и при прохождении электрического тока и при протека-нии различных процессов в их объёме.
Представление о том, что в растворах электролитов существуют свободные заряженные частицы – ионы, не сразу утвердилось в электро-химии. Первая теоретическая модель, объяснявшая явление электропроводности в проводниках второго рода была предложена Т. Гротгусом в 1805 г. применительно к процессу электрохимического разложения воды на водород и кислород. Пред-ставив молекулы воды в виде диполей, Гротгус располагал их цепочкой между катодом и анодом электролизера (рис. 1.1).
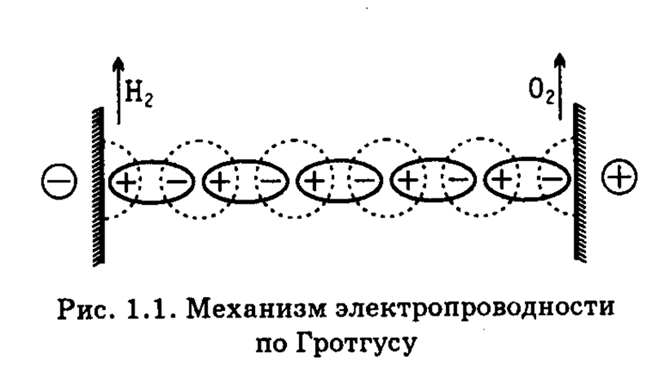
Рис.1.1. Механизм электропроводности по Гротгусу
Далее он предполагал, что при электролизе положительный конец диполя воды, обращенный к катоду, отщепляется и из него образуется водород. Аналогичным образом происходит отщепление отрицательного конца диполя, обращенного к аноду, с образованием кислорода.
После этого происходит перегруппировка положительных и отрицательных диполей в цепи, как это показано пунктиром на рис. 1.1.
Получающаяся при этом новая цепь диполей оказывается ориентированной против внешнего электрического поля, а потому диполи переориентируются. После пополнения цепочки диполей за счет молекул воды из объёма раствора не заключённого между электродами, процесс повторяется. Хотя, как выяснилось впоследствии, такой «эстафетный» механизм электропроводности имеет много общего с механизмом переноса тока ионами Н3О+ и ОН–, для большинства электролитов он оказался неприменимым.
Существенный шаг к современному представ-лению о строении растворов электролитов был сделан Фарадеем в 30-х годах Х1Х в. Фарадей одним из первых указал на возможность диссоциации электролита на ионы. Однако, по мнению Фарадея, это явление происходит только под влиянием электрического поля. В дальнейшем оказалось, что представления Фарадея об обра-зовании ионов под действием электрического поля оправдываются в растворах слабых электролитов при очень больших напряженностях поля. Так, например, при напряженности поля Х = 50 МВ/м диссоциация слабого электролита становится практически полной. Однако частично или полностью диссоциация электролита в растворе происходит и без всякого наложения поля. Об этом свидетельствуют следующие группы явлений обнаруженные различными исследователями при изучении растворов электролитов.
1. Осмотическое давление. Как следует из теории растворов, в достаточно разбавленных растворах осмотическое давление π связано с молярной концентрацией уравнением
π = сRT, (1.1.1)
где R – газовая постоянная; T – абсолютная температура.
Для растворов неэлектролитов, например, для раствора сахара в воде, уравнение (1.1.1) хорошо согласуется с экспериментальными данными. Для растворов электролитов, например NaCl, экспери-ментальное значение π оказывается существенно больше рассчитанного по уравнению (1.1.1). Для формального учета этого явления был введён изотонический коэффициент Вант-Гоффа і и соотношение для осмотического давления в раст-ворах электролитов записывали следующим образом:
π = ісRT, (1.1.2)
где і > 1.
Сопоставление формул (1.1.1) и (1.1.2) натал-кивало на мысль, что общее число частиц в растворах электролитов больше, чем в растворах неэлектролитов той же молярной концентрации.
2. Давление пара над раствором. Растворенное вещество снижает давление паров жидкости над раствором по сравнению с чистым растворителем на величину
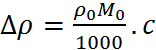 , (1.1.3)
, (1.1.3)
где р 0 – давление пара над чистым раство-рителем, М 0 – молекулярная масса растворителя.
Как и в опытах по осмотическому давлению, экспериментальные значения Δ ρ в растворах электролитов заметно превышают рассчитанные по формуле (1.1.3), в то время как для растворов неэлектролитов наблюдается хорошее согласие теории и опыта. Предположение о большем числе частиц в растворе электролита, т.е. введение в правую часть формулы (1.1.3) коэффициента і > 1, позволило устранить это противоречие.
3. Криоскопические и эбулиоскопические явления. Добавление растворенного вещества вызывает понижение температуры замерзания (Δ Тзам) и повышение температуры кипения (Δ Ткип) раствора по сравнению с чистым растворителем. В достаточно разбавленных растворах величины Δ Тзам и Δ Ткип можно связать с числом растворенных частиц:
∆ Tзам = 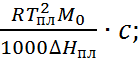 (1.1.4)
(1.1.4)
∆ Tкип = 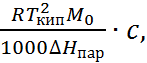 (1.1.5)
(1.1.5)
где Tпл и Ткип – температуры плавления и кипения растворителя: Δ Нпл и Δ Нпар – скрытые теплоты его плавления и парообразования.
Измерения температуры замерзания и температуры кипения растворов электролитов показали, что Δ Тзам и Δ Ткип заметно превышают рассчитанные по формулам (1.1.4) и (1.1.5), а также экспериментальные данные для растворов неэлектролитов тех же молярных концентраций.
4. Тепловой эффект реакции нейтрализации. Тепловой эффект реакции нейтрализации сильной кислоты сильным основанием в разбавленных растворах практически не зависит от химической природы кислот и оснований. Например, для двух реакций
HCl + NaOH → NaCl + H2O + Δ Н 1 (1.1.A)
HNO3 + KOH → KNO3 + H2O + Δ Н 2 (1.1.Б)
изменения энтальпии одинаковы: Δ Н 1 = Δ Н 2 = −57.3 кДж/моль (при 20 0С), хотя природа реагирующих веществ и продуктов реакции совершенно различна (за исключением получаю-щейся в результате обеих реакций воды). В тоже время представление о диссоциации кислот и оснований позволяет свести реакции (1.1.A) и (1.1.Б) к одному и тому же процессу Н+ + ОН- → H2O, который сопровождается вполне опреде-ленным тепловым эффектом.
5. Корреляция между каталитическим действием кислот и их электропроводностью. Чем больше электропроводность кислоты при данной концентрации, тем более сильный каталитический эффект она оказывает на процесс гидролиза сложных эфиров. Этот параллелизм нельзя было объяснить с точки зрения теории Фарадея, поскольку согласно этой теории электропро- водность связана с возникновением ионов под действием электрического поля, тогда как гидролиз эфиров исследуется в отсутствии поля. С другой стороны, при допущении о самопроизвольной диссоциации кислот и электропроводность раствора кислоты, и ее каталитическое действие можно связать с одним общим фактором – концентрацией ионов водорода.
Основные положения теории Аррениуса
Развитие теоретических представлений о строе-нии растворов началось с теории электро-литической диссоциации Аррениуса. В 1887 г. сформулированы следующие основные положения.
1. При растворении молекулы неорганических и органических кислот, оснований и солей спонтанно диссоциируют на ионы, например:
HCl ↔ H+ + Cl–;
NaOH ↔ Na+ + OH–;
NaCl ↔ Na+ + Cl–;
CF3COOH ↔ CF3COO– + H+;
K2SO4 ↔ 2K+ + SO4–2.
Ионы представляют собой заряженные частицы, которые состоят из отдельных атомов, или из группы атомов. Предполагалось, что ионы ведут себя подобно молекулам идеального газа, т. е. не взаимодействуют друг с другом. Физические причи-ны, которые приводят к диссоциации электролитов, в теории Аррениуса не рассматривались. Не обсуждался также и вопрос о том, почему заряженные частицы, на которые должны были распространяться законы электростатики, не взаи-модействуют друг с другом в растворах.
2. Диссоциация молекул на ионы является неполной, т.е. не все молекулы электролита, а лишь некоторая их доля α, названная степенью диссо-циации, распадается на ионы; доля молекул, равная (1 - α), остаётся недиссоциированной. Таким обра-зом, если при диссоциации одной молекулы элек-тролита образуется v ионов, то концентрация ионов в растворе оказывается равной v 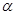 с, а концентрация недиссоциированных молекул - (1 - α) с. Следовате-льно, общая молярная концентрация частиц в растворе составит
с, а концентрация недиссоциированных молекул - (1 - α) с. Следовате-льно, общая молярная концентрация частиц в растворе составит
(1 -α) с + v с = с [1 + α(v – 1)].
Выражение [1 + α(v – 1)] показывает, во сколько раз увеличивается общая молярная концентрация частиц в растворе за счет диссоциации электролита, т.е. эквивалентно по своему физическому смыслу изотоническому коэффициенту Вант-Гоффа і. Поэтому по теории Аррениуса
і = 1 + α(v – 1). (1.2.1)
Поскольку v >1, а α > 0, то і > 1, и уравнение (1.2.1) позволяет дать разумное объяснение экспе-риментальным данным по осмотическому давле-нию, по изменению давления пара над растворами, а также по снижению температуры замерзания и по повышению температуры кипения растворов электролитов по сравнению с чистыми раство-рителями.
3. К процессу электролитической диссоциации применим закон действующих масс. Так, если в результате диссоциации молекул электролита МА получается один катион М+ и один анион А– (МА ↔ М+ + А–), то концентрации молекул и ионов равны соответственно:
[МА] = с (1 - α); [М+] = [А–] = α с,
и для константы электролитической диссоциации К по теории Аррениуса получаем следующее выражение:
K = 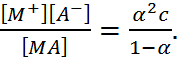 (1.2.2)
(1.2.2)
Поскольку обратная величина молярной концентрации V = 1/ с называется разведение, то уравнение (1.2.2) или аналогичное с заменой с = 1/ V называется законом разведения Оствальда. По теории Аррениуса константа К является постоян-ной для данного электролита. Поэтому по урав-нению (1.2.2) можно рассчитать степень диссоци-ации в зависимости от концентрации электролита. Решая квадратное уравнения и учитывая, что α > 0, получаем:
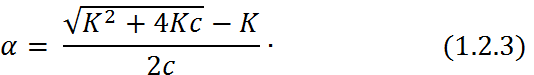
Как следует из уравнение (1.2.3), при условии
К ≫ 4 с α → 1, т. е. электролит становится полностью диссоциированным. С другой стороны, при малых константах диссоциации и при не очень низких концентрациях, когда К ≪ 4 с,
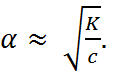 (1.2.4)
(1.2.4)
Соотношения (1.2.2) - (1.2.4) применимы только для растворов симметричных бинарных электролитов (т.е. если одна молекула электролита даёт один катион и один анион). Если же электролит имеет несимметричный валентный тип или имеется смесь электролитов, то математические соотношения, описывающие закон действующих масс согласно теории Аррениуса, и вытекающие из них следствия усложняются.
Теория Аррениуса позволила трактовать любые явления, связанные с ионными равновесиями 1), и легла, таким образом в основу качественного и количественного анализа.
1) В 1903 г. Сванте Аррениус за разработку этой теории был удостоен Нобелевской премии по химии.
На основе теории Аррениуса Я. Брёнстедом была сформулирована первая теория кислот и оснований, согласно которой кислотой (НА) является соединение, диссоциирующее на ионы водорода и кислотного остатка:
HА ↔ H+ + А–,
а основанием (МОН) – соединение, диссоции-рующее на катионы металла и анионы гидроксила:
МОН ↔ М+ + ОН–.
Таким образом, реакция нейтрализации сводится всегда к взаимодействию ионов H+ и ОН– и в разбавленных растворах сильных кислот и оснований должна по этому характеризоваться постоянством теплового эффекта независимо от природы кислоты и основания. Теория Аррениуса была широко применена к трактовке различных кислотно-основных равновесий. Для процессов диссоциации кислоты НА и основания МОН закон разведения Оствальда 2) можно записать в виде (1.3.1) (1.3.2), где К обозначает так называемую кажущуюся константу диссоциации соответственно кислоты (К А) или основания (К В)
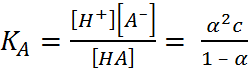 (1.3.1)
(1.3.1)
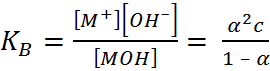 (1.3.2)
(1.3.2)
2) Вильгельм Оствальд применил представления Аррениуса к большой группе физико-химических явлений. В 1909 г. он был удостоен Нобелевской премии за работы в области катализа и развитие фундаментальных принципов управления химичес-кими равновесиями и скоростями реакций.
Так как 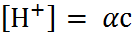 и
и 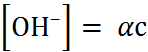 , то, используя уравнение
, то, используя уравнение
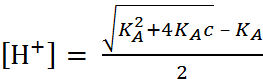 ; (1.3.3)
; (1.3.3)
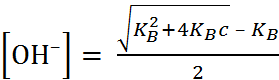 . (1.3.4)
. (1.3.4)
При 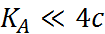 или
или 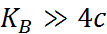 , т.е.
, т.е. 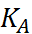 и
и 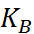 и не слишком малых c,
и не слишком малых c,
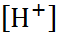 =
= 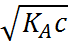 ; (1.3.5)
; (1.3.5)
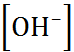 =
= 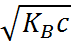 . (1.3.6)
. (1.3.6)
Логарифм концентрации ионов водорода, взятый с обратным знаком, называется pH
(С. Сёренсен):
pH = – lg 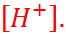 (1.3.7)
(1.3.7)
Cоответственно этому были выведены показатели диссоциации кислоты и основания:
p 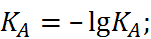 (1.3.8)
(1.3.8)
p  (1.3.9)
(1.3.9)
Применение теории Аррениуса к воде, которая относится к числу очень слабых электролитов и диссоциирует по уравнению
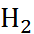 O
O 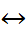
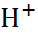 +
+ 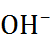 , позволяет записать:
, позволяет записать:
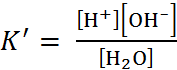 =
= 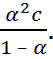 (1.3.10)
(1.3.10)
Так как для воды 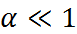 , а O практически постоянна, то получаем:
, а O практически постоянна, то получаем:
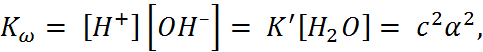 (1.3.11)
(1.3.11)
где 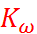 – константа, которая зависит от температуры и называется ионным произведе-нием воды.
– константа, которая зависит от температуры и называется ионным произведе-нием воды.
Ниже приведены значения p 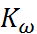 = – lg при различных температурах T:
= – lg при различных температурах T:
T, 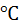 0 10 20 25 30 40 50 60
0 10 20 25 30 40 50 60
–lg 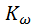 14,944 14,535 14,167 13,997 13,833 13,535 13,262 13,017
14,944 14,535 14,167 13,997 13,833 13,535 13,262 13,017
При 20 - 25 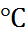 в первом приближении принимают p
в первом приближении принимают p 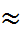 – 14. Из температурной зависимости p
– 14. Из температурной зависимости p 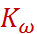 , воспользовавшись уравнением изохоры реакции, можно вычислить тепловой эффект диссоциации воды. При 20 он равен 57,3 кДж/моль, что практически совпадает по абсолютной величине с экспериментально найденной теплотой нейтрализации сильной кислоты сильным основанием в водных растворах, поскольку протекающая при этом реакция обратна процессу диссоциации воды.
, воспользовавшись уравнением изохоры реакции, можно вычислить тепловой эффект диссоциации воды. При 20 он равен 57,3 кДж/моль, что практически совпадает по абсолютной величине с экспериментально найденной теплотой нейтрализации сильной кислоты сильным основанием в водных растворах, поскольку протекающая при этом реакция обратна процессу диссоциации воды.
Соли, образованные слабыми кислотами или (и) слабыми основаниями, подвергаются в водных растворах гидролизу. Для соли слабой кислоты и сильного основания реакция гидролиза протекает по уравнению 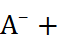 O
O 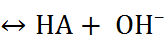 и характеризуется константой гидролиза
и характеризуется константой гидролиза
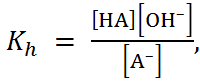 (1.3.12)
(1.3.12)
Поскольку 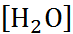 = const. Используя уравнения (1.3.1), (1.3.11) и (1.3.12) можно записать:
= const. Используя уравнения (1.3.1), (1.3.11) и (1.3.12) можно записать:
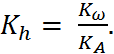 (1.3.13)
(1.3.13)
Уравнение (1.3.13) показывает, что константа гидролиза тем больше, чем слабее кислота.
На основе теории Аррениуса было сформулировано понятие о произведении растворимости 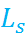 для труднорастворимых соединений типа
для труднорастворимых соединений типа 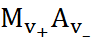
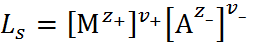 (1.3.14)
(1.3.14)
и объяснено уменьшение растворимости при добавлении в раствор веществ, имеющих общий ион с (заряды ионов 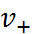 и
и 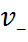 тривиаль-ными стехиометрическими соотношениями). Иногда это правило нарушается вследствие образования комплексных соединений. Так, растворимость цианида серебра AgCN повышается в присутствии цианида калия, так как при этом образуется ион
тривиаль-ными стехиометрическими соотношениями). Иногда это правило нарушается вследствие образования комплексных соединений. Так, растворимость цианида серебра AgCN повышается в присутствии цианида калия, так как при этом образуется ион 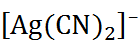 и устанавливается равновесие
и устанавливается равновесие
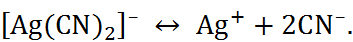
В случае одного вида лигандов реакцию комплексообразования можно представить в виде
q 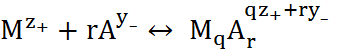 и ввести константу устойчивости комплекса
и ввести константу устойчивости комплекса 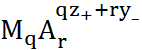 :
:
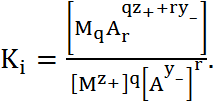 (1.3.15)
(1.3.15)
Часто комплексообразование протекает ступенчато:
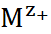 +
+ 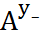 M
M 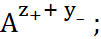
M 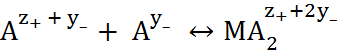
и т. д., причем каждая из таких стадий может быть охарактеризована константой устойчиво-сти.
Существует группа соединений, в молекулах которых содержатся кислотные и основные груп-пы. Такие соединения называются амфотер-ными электролитами, или амфолитами. Класси-ческий пример амфолитов – аминокислоты жирного ряда N 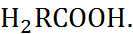 В водном растворе аминокислот в результате внутренней ионизации образуются цвиттер-ионы (двойные или биполярные ионы, амфиионы):
В водном растворе аминокислот в результате внутренней ионизации образуются цвиттер-ионы (двойные или биполярные ионы, амфиионы):
+NH2RCOOH 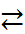 +NH3RCOO–
+NH3RCOO–
При добавлении сильной кислоты в раствор аминокислоты происходит реакция
+NH3RCOO– + H+ 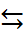 +NH3RCOOH,
+NH3RCOOH,
а при добавлении сильной щелочи – реакция
+NH3RCOO– + OH– NH2RCOO– + H2O.
Таким образом, основные свойства аминоки-слоты обусловлены присутствием группы –СОО–
а кислотные – группы –N 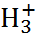 .
.

