 2015-08-21
2015-08-21 12343
12343Реакции в растворах часто ускоряются в присутствии веществ, относящихся к разновидностям кислот и оснований, названных именами Аррениуса, Льюиса, Бренстеда.
По общепринятому определению, данному в 1884 г. Сванте Аррениусом, кислота – соединение, дающее при диссоциации в воде катионы Н+, а основание – анионы ОН–.
По Д. Бренстеду и Т. Лоури (1923 г.) кислота – вещество, являющееся донором протона, а основание – акцептором протона.
Кислоту и основание, связанные уравнением типа (7.1), называют сопряженными
НА ↔ Н+ + А–. (7.1)
кислота протон основание
Протон в растворе обычно соединяется с молекулой растворителя, например с водой (7.2):
НА + Н2О ↔ Н3О+ + А–. (7.2)
Если молекулы растворителя не присоединяют и не отдают протоны, то растворенное вещество не проявляет ни кислотных, ни основных свойств, а сам растворитель называют апротонным.
Раствор кислоты в воде – пример системы, состоящей из двух пар сопряженных кислот и оснований, находящихся в равновесии (7.3):
НА + Н2О ↔ Н3О+ + А–. (7.3)
кислота основание кислота основание
По Гилберту Льюису (1938 г.) кислота – это акцептор неподеленной пары электронов, а основание – вещество, являющееся донором неподелённой пары электронов, которая может быть использована для образования устойчивой электронной конфигурации другого атома. Например, в реакции (7.4):
SO3 + H2O ↔ H2SO4, (7.4)
где H2O – основание, так как обладает свободной электронной парой, а SO3 – кислота, так как использует эту электронную пару.
Примерами кислот Льюиса являются такие соединения, как AlCl3, FeCl3, CuCl2, AlBr3, BF3. Примеры оснований Льюиса:NH3, N2H4, NO3–, C6H6, C6H5N.
Теория Льюиса гораздо шире, чем предыдущие теории, но имеет некоторые недостатки:
– сомнительно толкование протонных кислот, таких как H2SO4, НСl и других, так как они не могут присоединять электронную пару с образованием ковалентных связей;
– несостоятельность в определении силы кислот, поскольку по Льюису, это зависит от особенностей реакции.
Каталитические реакции, ускоряемые кислотами и основаниями, представлены широким перечнем химико-технологических процессов, таких как алкилирование парафинов и аренов олефинами, гидратация алкенов и алкинов, этерификация и гидролиз сложных эфиров, полимеризация олефинов в газовой фазе. Реакции, катализируемые кислотами и основаниями, можно разделить на три вида:
– специфический кислотный или основной катализ,
– общий кислотный или основной катализ,
– электрофильный или нуклеофильный катализ.
7.1.1. Специфический кислотный катализ
В этом случае катализаторами являются кислоты Аррениуса (ионы Н3О+), и реакция идет по схеме (7.5):
S + Н3О+ ↔ SH+ + H2O → P + Н3О+, (7.5)
где S – молекула субстрата,
P – молекула продукта.
Первая стадия – внедрение протона в реагирующую часть молекулы субстрата – протекает быстро. Вторая стадия, в которой образовавшийся катион SН+ отщепляет протон с образованием продукта, протекает медленно и является лимитирующей стадией процесса.
По этому механизму протекают реакции гидролиза сложных эфиров и ацеталей, гидратации ненасыщенных альдегидов, дегидратации третичных спиртов, кетоенольной изомеризации.
7.1.2. Специфический основной катализ
Катализаторами здесь являются основания Аррениуса – гидроксид-ионы ОН–. Реакция протекает по схеме (7.6):
SH + ОН– ↔ S– + H2O → S* + OH– → P + OH–, (7.6)
где S* – новое промежуточное соединение, причем первая стадия быстрая, а вторая – медленная, то есть лимитирующая весь процесс.
По этому механизму протекают реакции гидролиза сложных эфиров, гидратации альдегидов, альдольной конденсации, реакции, включающие перегруппировки Кляйзена, Михаэля, Перкина.
7.1.3. Общий кислотный катализ
Он отличается от специфического тем, что здесь катализаторами процесса служат кислоты Бренстеда (НА), то есть ион Н3О+ не является донором процесса, и реакция идет по схеме (7.7):
S + HA → SH+ + A– → P + HA. (7.7)
Здесь медленной стадией является образование иона SH+, то есть лимитирующей является первая стадия процесса.
По этому механизму протекают реакции кетоенольной изомеризации, присоединения к карбонильной и карбоксильной группе.
7.1.4. Общий основной катализ
Катализаторами процесса выступают основания Бренстеда (В), то есть акцептором протона не является анион OH–. Процесс описывается схемой (7.8):
SH + B → S– + BH+ → S* + B → P + B. (7.8)
Самой медленной стадией является образование активного аниона.
По этому механизму идут реакции гидролиза эфиров, конденсации альдегидов.
7.1.5. Электрофильный катализ
В этом случае катализаторами служат кислоты Льюиса. Объяснение роли кислот Льюиса как катализаторов связывают с образованием ими за счет донорно-акцепторной связи промежуточного соединения с одним реагентом, которое более легко вступает в реакцию с молекулами второго реагента благодаря наличию областей с повышенной или пониженной электронной плотностью.
По этому механизму протекают реакции Фриделя–Крафтса, гидролиза сложных эфиров аминокислот в присутствии катионов переходных металлов.
7.1.6. Кинетика реакций кислотно-основного катализа
Рассмотрим наиболее общий случай, когда реакция катализируется одновременно кислотами и основаниями Аррениуса (Н3О+ и ОН–).
Механизм такой реакции можно представить уравнениями (7.9 – 7.12):
SH + H3O+  HSH+ + H2O (7.9)
HSH+ + H2O (7.9)
SH + OH–  S– + H2O (7.10)
S– + H2O (7.10)
SH + H2O  HSH+ + OH– (7.11)
HSH+ + OH– (7.11)
SH + H2O  S– + H3O+ (7.12)
S– + H3O+ (7.12)
При этом реагент SH участвует одновременно во всех четырех реакциях, отвечающих разным типам катализа.
Скорость расхода реагента SH запишем следующим образом (7.13):
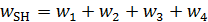 (7.13)
(7.13)
или
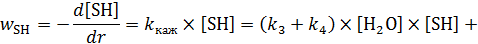
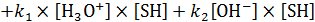 . (7.14)
. (7.14)
Сделаем некоторые допущения:
1) 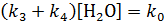 , где k 0– константа скорости реакции реагента с водой (некаталитическая стадия);
, где k 0– константа скорости реакции реагента с водой (некаталитическая стадия);
2) 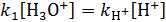 , где
, где  – константа скорости реакции, катализируемой протонами;
– константа скорости реакции, катализируемой протонами;
3) 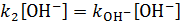 , где
, где 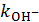 – константа скорости реакции, катализируемой ионами гидроксила.
– константа скорости реакции, катализируемой ионами гидроксила.
Таким образом, кажущаяся константа скорости реакции примет вид (7.15):
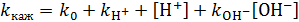 . (7.15)
. (7.15)
Так как 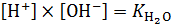 , то выражение (7.15) можно записать в виде (7.16):
, то выражение (7.15) можно записать в виде (7.16):
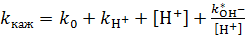 , (7.16)
, (7.16)
где 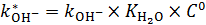 ,
,
С 0 – стандартная концентрация.
Уравнение (7.16) позволяет рассмотреть все случаи кислотно-основного катализа. Графики зависимости lg [ k каж] = f (pH) для каждого случая приведены на рисунке 7.1.
Наиболее общий случай (а) изображен линией, которая указывает на три различные области рН, характеризуемые прямыми отрезками, наклон которых к оси абсцисс составляет – 1; 0 и + 1. В зависимости от величины рН преобладает либо специфический кислотный, либо основной катализ, а в промежуточной области – каталитическое действие воды. По данному типу катализа протекает реакция мутаротации глюкозы.
В случае (см. рис. 7.1, линия b) каталитическое действие воды происходит очень медленно по сравнению с реакциями, катализируемыми ионами Н+ и ОН–. Существует два отрезка прямой (горизонтальный участок кривой отсутствует), которые пересекаются в точке с минимальным значением k каж. Этому типу катализа отвечает реакция галогенирования ацетона.
Кривая с на рисунке 7.1 соответствует реакциям, катализируемым только ионами ОН– в растворах, когда рН > 7.
Кривая d представляет случай, когда реакция всегда катализируется ионами ОН–.
Кривая e описывает реакцию, катализируемую ионами Н+ в растворах, когда рН имеет достаточно малые значения.
Кривая f характеризует пример, в котором реакция всегда катализируется ионами Н+.
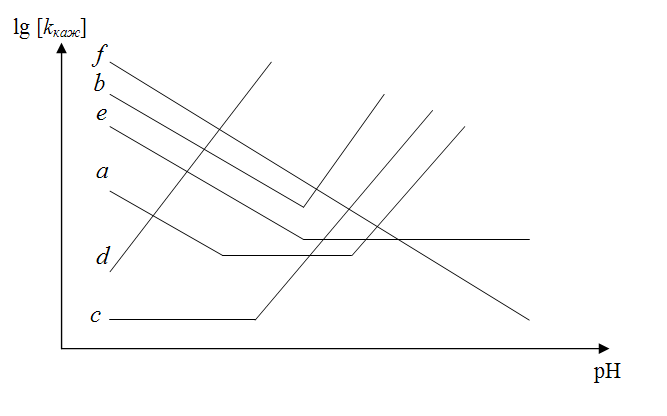
Рис. 7.1. Логарифмическая зависимость кажущейся константы
скорости реакций, катализируемых кислотами и/или основаниями

