 2015-02-14
2015-02-14 2260
2260Различают газо-адсорбционную (газо-твердофазную) хроматографию (ГАХ), где неподвижной фазой является твердофазный сорбент, и газо-жидкостную (ГЖХ), в которой неподвижной фазой служит слой жидкости (жидкая пленка) на поверхности твердого носителя. Подвижной фазой в обоих видах хроматографии является газ-носитель и пары разделяемых веществ, поэтому разделяемые компоненты перед вводом в хроматографическую колонку переводят в испарителе в парообразное состояние, а затем с потоком газа-носителя напрявляют в колонку. В качестве газа-носителя используют инертные газы (азот, гелий, аргон) высокой степени очистки.
В ГАХ применяют различные адсорбенты (силикагель, уголь, оксид алюминия) с высокой удельной поверхностью, и распределение веществ между неподвижной и подвижной фазами определяется процессом адсорбции, т.е. происходит концентрирование веществ на поверхности раздела фаз за счет сил межмолекулярных взаимодействий, имеющих электростатическую природу.
В ГЖХ распределение компонентов между подвижной и неподвижной фазами основано на различии их растворимости в жидкой фазе.
В аналитической практике ГЖХ применяется чаще из-за большого разнообразия жидких фаз, обеспечивающих эффективное разделение веществ в достаточно широком интервале концентраций.
Принципиальная схема выполнения газовой хроматографии состоит в следующем. Сначала микрошприцем вводят в испаритель индивидуальные вещества (стандарты), наличие которых предполагается в анализируемой смеси, а затем при тех же условиях вводят пробу анализируемой смеси. Пары веществ с потоком инертного газа-носителя проходят через колонку с поглотителем (адсорбентом) и поглощаются в колонке, а газ-носитель свободно выходит из колонки и попадает на детектор. Через некоторое время, называемое временем удерживания, из колонки вместе с газом-носителем выходят (десорбируются) и поглощенные адсорбентом компоненты и так же попадают на детектор. В качестве детектора чаще других применяют детектор на основе изменения теплопрододности газовых потоков (катарометр). Катарометр состоит из двух ячеек (камер), в каждой из которых находится по плечу моста сопротивлений при строго постоянной и одинаковой температуре. Через камеру сравнения газ-носитель проходит до входа в испаритель, а через рабочую измерительную камеру – непосредственно после выхода из колонки. Когда в колонку ничего не введено, через обе камеры проходит чистый газ-носитель, газовый состав в обеих камерах одинаковый, теплопроводность и сопротивления плеч моста тоже одинаковые, поэтому мост сопротивлений находится в сбалансированном состоянии и регистрируемый ток равен нулю. При вводе в колонку индивидуальных компонентов или анализируемой смеси до тех пор, пока поглощенные вещества удерживаются на неподвижной фазе, через обе камеры, по-прежнему, идет только чистый газ-носитель и регистрируемый ток равен нулю. Через некоторое время (время удерживания) газ-носитель начинает десорбировать компоненты с поглотителя в рабочую (измерительную) камеру детектора. Газовый состав в камерах становится неодинаковым, мост сопротивлений выходит из баланса и в системе регистрации появляется ток. Когда компонент полностью выходит в рабочую камеру, ток достигает максимальной величины. На хроматограмме регистрирующего устройства (ленте самописца или дисплея компьютера) появляются соответствующие пики (рис. 5.2). Принципиальная схема газового хроматографа приведена на рис. 5.8.
Рисунок 5.8 – Принципиальная схема газового хроматографа: 1 – баллон с газом-носителем; 2 – редуктор; 3 – чистый газ-носитель; 4 – камера сравнения детектора-катарометра; 5 – измерительная (рабочая) камера катарометра); 6 – вывод газа из хроматографа; 7 – детектор; 8 – газ-носитель; 9 – устройство (дозатор) для введения парообразных анализируемых веществ в хроматографическую колонку; 10 – спиральная колонка с сорбентом; 11 – термостат; 12 – регистрирующее устройство; 13 – мост сопротивлений (мост Уитстона); 14 – ротаметр
При благоприятном сочетании химической природы разделяемых компонентов, адсорбента-поглотителя и газа-носителя компоненты имеют различные значения времени удерживания и хорошо разделяются. На хроматограмме получается хорошее разрешение пиков RS.
Качественная идентификация компонентов анлизируемой смеси осуществляется путем сравнения времени удерживания t R (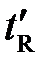 , t отн.) компонентов со временем удерживания известных веществ-идентификаторов на хроматограммах, полученных при одинаковых условиях хроматографирования.
, t отн.) компонентов со временем удерживания известных веществ-идентификаторов на хроматограммах, полученных при одинаковых условиях хроматографирования.
При количественных определениях, как уже отмечалось, используют, главным образом, два метода.
Метод нормировки применяют для определения относительного содержания компонентов в разделяемой смеси по отношению площади пика компонента на хроматограмме Sx к сумме площадей всех пиков:
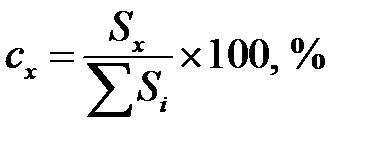 или
или 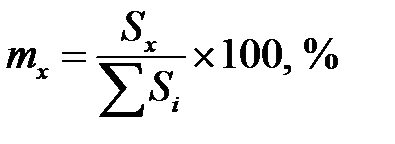 . (5.9)
. (5.9)
Если чувствительность детектора к компонентам смеси различная, то в эти уравнения вносят поправочные коэффициенты fi (см. уравнение 5.7).
Площади пиков рассчитывают как площади треугольника (S = hμ /2). Если пик очень острый или, наоборот, форма пика лишь приближенно соответствует треугольнику, то условную площадь пика находят как произведение времени выхода компонента на высоту пика в максимальной точке (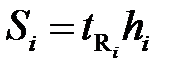 ). И тогда:
). И тогда:
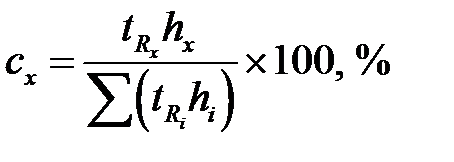 .
.
Для определения абсолютного содержания компонентов применяют метод внешних стандартов. Приготавливают два (или более) раствора определяемого компонента с разной, но известной концентрацией. Затем поочередно вводят строго одинаковые объемы стандартов и пробы в хроматограф. На полученных хроматограммах измеряют площади пиков стандартов и компонента пробы и одним из методов определяют неизвестную концентрацию компонента в пробе. Чаще всего используют метод градуировочного графика (рис. 5.5) или метод сравнения: 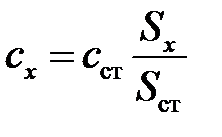 , где с ст и S ст – концентрация раствора стандарта и его площадь пика на хроматограмме, соответственно; сх и Sx – соответственно, концентрация и площадь пика аналита.
, где с ст и S ст – концентрация раствора стандарта и его площадь пика на хроматограмме, соответственно; сх и Sx – соответственно, концентрация и площадь пика аналита.
Литература
1. Основы аналитической химии. В 2-х кн. Учебник для вузов / Под ред. Академика Ю.А. Золотова. 3-е изд. перераб. и доп. Кн. 1 Общие вопросы. Методы разделения. М.: Высшая школа, 2004, 361 с.; Кн. 2 Методы химического анализа. М.: Высшая школа, 2004, 503 с.
2. Аналитическая химия. В 3-х т. Учебник для вузов под ред. проф. Москвина Л.Н. Т. 1. Методы идентификации и определения веществ (Белюстин А.А., Булатов М.И., Дробышев А.И. и др.). М., Изд. центр «Академия», 2008, 576 с.; Т. 2. Методы разделения веществ и гибридные методы анализа (Зенкевич И.Г., Карцова Л.А., Москвин Л.Н. и др.). М.: Изд. центр «Академия», 2008, 304 с.
3. Васильев В.П. Аналитическая химия. В 2-х кн. Кн. 2 Физико-химические методы анализа. М.: Дрофа, 2004, 367 с.
4. Жуков А.Ф., Колосова И.Ф., Кузнецов В.В. и др. Аналитическая химия. Физические и физико-химические методы анализа. Учебник для вузов. Под ред. О.М. Петрухина. М.: Химия, 2001, 496 с.
5. Пилипенко А.Т., Пятницкий И.В. Аналитическая химия. В 2-х кн. Кн. 1 – М.: Химия, 1990, 480 с.
6. Алесковский В.Б., Бардин В.В., Булатов М.И. и др. Физико-химические методы анализа. Практическое руководство / Под ред. В.Б. Алесковского. Л.: Химия, 1988, 376 с.
7. Булатов М.И., Калинкин И.П. Практическое руководство по фотометрическим методам анализа. Л.: Химия, 1986, 432 с.
8. Беляев А.П., Рубец В.П., Зарембо В.И. Феноменология и практика обработки эксперимента в аналитической химии (физико-химические методы анализа). Учебное пособие. СПб.: Изд-во СПбГТИ(ТУ), 1999, 110 с.
9. Булатов М.И., Маметнабиев Т.Э. Фотометрические методы анализа. Учебное пособие. СПб.: Изд-во СПбГТИ(ТУ), 2008, 92 с.

