 2015-06-10
2015-06-10 1341
1341Состояние, именуемое синдромом дыхательных расстройств взрослых, а за рубежом называемое респираторным дистресс-синдромом взрослых (ARDS), было описано D. G. Ashbaugh и соавт. (1967) на основе изучения данных о 12 больных, у которых клиническая картина заболевания характеризовалась одышкой, гипоксемией, сниженной податливостью легких, диффузными альвеолярными инфильтратами и интерстициальным отеком легких. Авторы предложили и ввели термин ARDS (в котором фигурирует слово «взрослых»), полагая, что дефект сурфактантной системы при описываемом синдроме сходен с тем, который бывает при специфическом синдроме дыхательных расстройств новорожденных. В дальнейшем F. D. Moore (1969) показал, что феномен повреждения сурфактантной системы вторичен и не является ведущим в патогенезе синдрома. F. D. Moore описал синдром как неспецифическое следствие шока и травмы и других инициирующих факторов, например передозировки инфузионных сред, переливания крови, жировой эмболии и др. Справедливости ради надо сказать, что СДРВ, поразивший воображение D. G. Ashbaugh и соавт. и надолго приковавший к себе внимание в 70—80-х годах, описывался и ранее под различными наименованиями. Хорошо известны сейчас термины «шоковое легкое», «влажное легкое», «тяжелое легкое», «легкие Дананга», «некардиогенный отек легких», «болезнь гиалиновых мембран взрослых» и др.
В клинической практике наиболее часто наблюдаются варианты ОДН, возникающей у крайне тяжело больных, перенесших стрессовую ситуацию, например тяжелую травму, кровопотерю, большую операцию с гемотрансфузией или массивной инфузией растворов, или находящихся в критическом состоянии в результате сепсиса (септический шок), панкреатита, перитонита, гиповолемии. В течение последних десятилетий СДРВ стал предметом особого внимания специалистов различного профиля, прежде всего реаниматологов, а также хирургов, кардиологов, пульмонологов. В связи с этим значительно улучшилась диагностика синдрома, врачи стали чаще ставить обоснованный диагноз начальных форм ОДН даже при сохраненной функции внешнего дыхания и в отсутствие явных признаков гипоксии.
По данным F. D. Moor и соавт. (1969), N. O'Connor и F. D. Moor (1980), СДРВ наблюдался у 7з больных, умерших после тяжелых хирургических заболеваний.
В начале 70-х годов в США были проведены исследования по национальной программе изучения и лечения ОДН. По данным 9 отделений интенсивной терапии, оценено 150000 больных с ОДН, поступивших в течение года. 40 000 больных из этого числа умерли непосредственно от ОДН. Эти результаты указывают на чрезвычайную важность проблемы в целом.
W. R. Baumari и соавт. (1986) наблюдали 11 112 больных, поступивших в палаты интенсивной терапии. Из них 4926 больных находились в крайне тяжелом состоянии. Из этого числа у 90 пациентов (около 2% числа крайне тяжело больных) развился СДРВ. Смертность больных с СДРВ составила 64%. Особенно высокая смертность (86%) отмечена среди больных, у которых ОДН была обусловлена пневмоцистной пневмонией и аспирационным синдромом.
Сочетание СДРВ с поражением нескольких функций и систем одновременно обусловливает и высокую летальность этих больных [Гологорский В. А. и др., 1988]. По данным N. O'Connor и F. D. Moor (1980), летальность составляет 49%, если СДРВ является единственным заболеванием, но возрастает до 63%, если в процесс вовлекался еще один орган, до 91% —при поражении двух органов и до 97%, если, помимо легких, возникает недостаточность еще 3—4 органов. Как правило, это острая недостаточность функции почек, печени, сердечно-сосудистой системы, заболевания ЦНС или расстройства коагуляции крови. Летальность при СДРВ в значительной мере зависит от возраста: больные старше 65 лет умирают в 85% случаев. Таким образом, СДРВ можно определить как неспецифическое поражение легких, возникающее у крайне тяжело больных и приводящее к гипоксии.
С позиций патофизиологии синдром характеризуется нарушением диффузии газов и возникновением внутрилегочного сброса крови (шунта) справа налево [West J. В., 1990], с диагностических— гипоксией и зависимостью больного от высокой концентрации О2 во вдыхаемой смеси, наконец, с лечебных позиций — неизбежностью интубации трахеи (трахеостомия, ИВЛ и необходимость применения сложного комплекса лечебных мероприятий).
В качестве первичной причины СДРВ на первом месте находится сепсис. По данным R. L. Fulton и С. Е. Jones (1975), СДРВ развился у 44 из 399 больных с тяжелой травмой. У 40 из них непосредственной причиной СДРВ был сепсис. Следовательно, лечение самого сепсиса является важнейшей мерой профилактики СДРВ.
Клиническая картина. - Принято различать четыре фазы -СДРВ Описаны симптомы и лабораторные данные в каждой из них [Moore F. D. et al., 1969].
Первая фаза (фаза повреждения, или ранняя обратимая фаза) начинается обычно немедленно после эпизода агрессии — травмы, ожога, операции, кровопотери, инфаркта миокарда, септического шока и т. д. В ряде случаев эта фаза клинически не проявляется и не имеет дальнейшего развития. При дальнейшем развитии ОДН наиболее характерным клиническим признаком становится умеренная гипервентиляция (одышка), которую больной чаще всего легко переносит.
Важным фактором в возникновении критического состояния являются различные варианты расстройства кровообращения. При низкой объемной скорости кровообращения, даже в отсутствие газообменных нарушений, организм страдает от гипоксии, которая в таких случаях может быть гемической (если произошла кровопотеря) или циркуляторной. Происходит интенсивное накопление метаболических кислот, особенно лактата, сначала в тканях, а затем в крови. Если после первичного нарушения гемодинамики кровообращение улучшается, то довольно быстро восстанавливается метаболизм лактата или происходит выведение последнего — развивается метаболический алкалоз. В ряде случаев алкалоз развивается как гипокалиемический. Наблюдающаяся в этом периоде у большинства больных спонтанная гипервентиляция приводит к гипокапнии (РаСо, ниже 33 мм рт. ст.) и обусловливает дыхательный алкалоз. Таким образом, наиболее характерными признаками первой фазы ОДН у больных в критическом состоянии являются спонтанная гипервентиляция и метаболический алкалоз. Нередко больной благополучно переживает первичные циркуляторные расстройства и дыхание восстанавливается без выраженных повреждений легких.
Больные в критическом состоянии обычно подвергаются интенсивному лечению. В число основных мероприятий входят инфузии различных жидкостей, белковых растворов, плазмы, иногда (по специальным показаниям) гемотрансфузии и др. Некоторые элементы подобной интенсивной терапии могут служить основой последующих расстройств легочных функций. Если в ходе лечения не удается быстро получить желаемый эффект восстановления кровообращения и возникает необходимость продолжать массивную инфузионную терапию, то наиболее вероятно дальнейшее развитие дыхательных расстройств, приводящих к гипоксии.
Вторая фаза (ранняя прогрессирующая). Дальнейшее прогрессирование СДРВ занимает от нескольких часов до нескольких дней. Усиление одышки происходит на фоне стабильного, иногда удовлетворительного общего состояния больного и в связи с этим кажется необъяснимой. У больных в крайне тяжелом состоянии, а также при интенсивном лечении, включающем массивные гемотрансфузии, дыхательные расстройства проявляются в ранние сроки. Более позднее развитие дыхательных расстройств характерно для септических состояний и после токсико-инфекционного шока, острого панкреатита или при продолжающемся перитоните.
Довольно длительно, иногда в течение нескольких суток, кроме одышки, не удается обнаружить никаких других клинических признаков прогрессирующего СДРВ. При тщательном обследовании — физикальном и рентгенологическом — не выявляется никаких отклонений от нормы. Однако при исследовании газового состава артериальной крови обнаруживается снижение Ро2 до 75—70 мм рт. ст. при дыхании комнатным воздухом. Проба с дыханием 100%, О2 указывает на недостаточное повышение Рао2 и увеличение легочного шунта в этой фазе да 10—15% минутного объема сердца (норма — не более 2 —3% сердечного выброса). Таким образом, уже во второй фазе значительная часть крови, протекающей по легким, не оксигенируется. Следовательно, эта фаза характеризуется развитием гипоксемии. Вместе с тем, несмотря на прогрессирующую ОДН, состояние больного остается удовлетворительным.
Третья фаза (поздняя прогрессирующая) характеризуется дальнейшим ухудшением состояния больного и нарастанием, явных признаков ОДН. Одышка становится выраженной, иногда мучительной, дыхательный объем увеличивается в 1,5—2раза против нормы, нарастает гипоксемия в сочетании с гипокапнией. Наиболее характерный клинический и патофизиологический феномен — зависимость больного от кислорода. Гипоксия приобретает стойкий характер. Обычно в этой стадии интубируют трахею и начинают ИВЛ. Легочный шунт достигает 20—30% минутного объема сердца. Однако если отсутствует бронхиальная обструкция, Р а о2 не возрастает.
К нарастающей гипоксемии присоединяются и другие тяжелые изменения: повышается бронхиальная секреция, возникают множественные спонтанные эмболии мелких легочных сосудов, бактериальная пневмония, развивается ДВС-синдром, катастрофически увеличивается водное переполнение легких. Аускультативно выявляются многочисленные сухие и влажные хрипы. Рентгенологически определяются очаговые и диффузные инфильтраты.
Ра02 удается поддерживать на близком к норме уровне лишь при помощи ингаляций О2 в высоких концентрациях. Величина рН крови чаще всего близка к норме из-за гипокапнии, относительно уравновешивающей нарастающую лактацидемию.
Таким образом, важнейшими признаками третьей фазы являются выраженные нарушения оксигенации крови в легких, а также неспособность больного поддерживать эффективную спонтанную вентиляцию легких.
Если осуществляется ИВЛ, то выживание больного в этой фазе все же возможно, хотя и является скорее исключением, чем правилом. Оно начинается с улучшения ответа организма да проводимую терапию, снижения зависимости больного от О2.
Вместе с отсутствием повышения Раo2 при ингаляции 100% О2 (проба Уленбрука) неблагоприятным прогностическим признаком является некупируемая гипоксемия и прогрессирующее увеличение концентрации лактата в крови. Со временем у больного развиваются кома и -последующее угасание спонтанной дыхательной активности. Наблюдающаяся ранее гипокапния «меняется повышением Рао2.
Четвертая фаза (терминальная) является финальной и кратковременной. Прогрессирует кома, еще большим становится легочный шунт (иногда 50—60% минутного объема сердца). Концентрации лактата и Расо2 увеличиваются до предельных значений. Нарастающий метаболический ацидоз (рН<7,15—7,10) не поддается коррекции. Оксигенация крови в этой фазе не улучшается с увеличением фракционной концентрации О2 во вдыхаемом газе (FiO2) и положительного давления в конце выдоха (ПДКВ), растяжимость легких все более снижается. Выживание больных в четвертой фазе СДРВ практически невозможно.
Клиническая картина дополняется нарушениями других функций и систем: прогрессируют гипотензия, олигурия, кома. Определяющие судьбу больного нарушения метаболизма имеют в своей основе не только гипоксемию, но и синдром низкого периферического кровотока. Обычно наблюдаемые аускультативные феномены и данные рентгенологического исследования в этой фазе мало помогают врачу правильно ориентироваться в ситуации. Прогрессирующая тяжелая легочная и генерализованная инфекция существенно ухудшает течение синдрома. На ЭКГ, можно видеть изменения, отражающие общую и субэндокардиальную ишемию миокарда. Обусловленное гипоксией и ацидозом замедление ритма может перейти в асистолию без каких-либо предшествующих проявлений.
Лабораторная диагностика. Развивающаяся в связи с одышкой гипокапния наблюдается обычно в течение второй и третьей фаз СДРВ. В зависимости от состояния бронхиальной проходимости Расо2. У больных колеблется в пределах 25—35 мм рт. ст.
С началом ИВЛ гипокапния становится более выраженной, так как возникает необходимость применять высокие объемы вентиляции легких. Выраженная гипокапния при СДРВ может быть опасной из-за возможности церебральной вазоконстрикции, смещения кривой диссоциации оксигемоглобина (КДО) влево и повышения возбудимости миокарда. Необходимо по возможности избегать этих вредных влияний гипокапнии. Если РаСО2 ниже 30—33 мм рт. ст., то нужно попытаться восстановить его до нормы увеличением объема мертвого пространства путем дополнительной вставки в систему дыхательных путей. Для претерминальных ситуаций характерно спонтанное восстановление Расо2 до нормальных величин, а затем медленное увеличение его до уровня выше нормы, несмотря на гипервентиляцию.
Расстройства КОС весьма характерны для СДРВ. Низкий сердечный выброс в начале заболевания вызывает увеличение концентрации лактата (L—) в крови до 2—5 ммоль/л. Таким образом, метаболический ацидоз при СДРВ почти целиком связан с накоплением L—. Как правило, L—определяется в форме «избытка лактата», т. е. накапливается в количестве, превышающем тот уровень, который может быть объяснен повышением содержания пирувата. При компенсированной ОДН, например, в первой и второй фазах СДРВ большая часть избытка лактата метаболизируется или зкскретируется и развивается средней выраженности метаболический алкалоз. Обычно алкалоз является результатом комбинации различных факторов, например потери НС1 при назогогастральной аспирации или после рвоты, инфузии растворов гидрокарбоната натрия в начальном периоде лечения, метаболизма стабилизатора донорской крови цитрата натрия при гемотрансфузиях. Алкалоз усиливается в связи с секрецией альдостерона, который способствует задержке NaHCO3 почками, особенно на фоне гипохлоремии. В терминальной фазе алкалоз вновь сменяется гипоперфузионным метаболическим ацидозом. Концентрация L— в крови повышается до уровня, выше наблюдавшегося в первой фазе; к моменту смерти он может достигать 30 ммоль/л или больше.
Другие лабораторные проявления у этих пациентов в целом определяются преобладающими в данный момент нелегочными синдромами, являющимися выражением полиорганной дисфункции, например почечной недостаточностью, поражением ЦНС, недостаточностью сердечно-сосудистой систем, печени и системы коагуляции крови. Почечная недостаточность ведет к повышению концентраций мочевины и креатинина в крови, повышению К+ и острой гипокальциемии. Снижается экскреция лекарств из организма, особенно мышечных релаксантов и антибиотиков.
В связи с развитием гипоксии возникают расстройства сознания вплоть до комы. На ЭЭГ часто регистрируются нарушения ритмики, однако ценность этого исследования в диагностике поражения ЦНС при ОДН невелика.
Острая печеночная недостаточность при критических состояниях наиболее часто развивается так же, как у септических больных, или вследствие системной гипоперфузии. В значительной степени этому способствует и гипоксия.
Увеличение активности печеночных ферментов, особенно щелочной фосфатазы, наблюдается, как правило, раньше, иногда задолго до гипербилирубинемии. Наконец, при ОДН нередко развиваются изменения свертывающей системы крови. Часто они бывают следствием массивной гемотрансфузии, обширного повреждения тканей или сепсиса. Существенно снижается количество тромбоцитов, а также укорачивается (а при поздних фазах удлиняется) протромбиновое и активированное частичное тромбопластиновое время, что может свидетельствовать о развитии ДВС-синдрома. Повышение уровня продуктов деградации фибрина и фибриногена (ПДФ) свидетельствует о глубоком расстройстве внутрисосудистой коагуляции и распаде образующихся в сосудах сгустков крови.
Рентгенологическая диагностика. Информативность рентгенологической картины при СДРВ в значительной степени зависит от выраженности отека легких и продолжительности самого синдрома. При этом на рентгенограммах можно видеть признаки венозной гипертензии, проявляющиеся расширением рисунка венозной сети преимущественно в области верхушек легких, в сочетании с умеренным спазмом вен в нижних отделах их.
Отек легких может развиваться как в интерстициальной ткани, так и внутриальвеолярно. Иногда ему сопутствуют гипернатриемия и высокая осмоляльность плазмы. Появление влажных хрипов в легких свидетельствует о накоплении жидкости в альвеолярном пространстве. Для успеха лечения чрезвычайно важно диагностировать начальные стадии отека. Рентгенологическое исследование позволяет сделать это довольно уверенно.
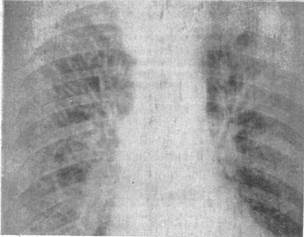
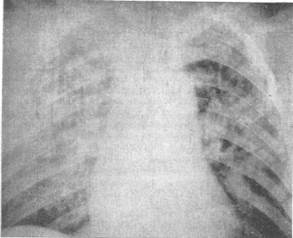
Рис. 4.1. Рентгенологическая картина при СДРВ.
а — начальные фазы синдрома: усиление сосудистого компонента легочного рисунка главным образом за счет венозных стволов, возникновение очаговоподобных тканей, периваскулярные и перибронхиальные муфты: б — понижение прозрачности легочного рисунка, сливающиеся очаговоподобные хлопьевидные тени различной выраженности.
Интерстициальный отек легких в начальных стадиях СДРВ может проявляться на рентгенограммах хлопьевидными размытыми тенями, усилением сосудистого компонента легочного рисунка, главным образом за счет венозных стволов, наличием горизонтально распространяющихся тонких линий преимущественно в краевых отделах легких (рис. 4.1, а).
При нарастании интерстициального отека легких на рентгеновских снимках сосудистый рисунок еще более усиливается, появляются сетчатость его и очаговоподобные тени. Отмечается также размытость теней сосудов. Вокруг сосудов, отображенных в срезе, возникают периваскулярные и перибронхиальные муфты.
При хорошем качестве рентгенограмм удается видеть набухшие, радиально расположенные лимфатические сосуды в виде тонких прямых линий, пересекающих крупные сосуды.
В поздних фазах рентгенологическая картина СДРВ характеризуется слиянием очаговоподобных теней и линий однообразной плотности, усилением рельефа сосудистого рисунка в области корня легкого и снижением прозрачности легочного рисунка (рис. 4.1,6). В результате образования легочного патологического шунта и в связи с тромбозом мелких и средних легочных сосудов возникают другие многочисленные тени неспецифической природы неодинаковых размеров и конфигураций, расположенные в различных областях легких, чаще в непосредственной близости к корню легкого.
Этиологические и патогенетические факторы. СДРВ — состояние, которое вызывается множеством причин и их сочетаний и, как правило, возникает в условиях критического состояния. Поскольку СДРВ сопутствует тяжелым повреждениям, пытались связать возникновение поздних дыхательных расстройств у раненых с прямой травмой грудной клетки и непосредственной травмой легких. Однако оказалось, что у раненых с обширной травмой тела, в частности с повреждениями нижних конечностей, СДРВ развивается чаще, чем у раненых с травмой грудной клетки. Создавалось впечатление, что главной причиной СДРВ является травматический или токсико-инфекционный шок. Дальнейшие исследования показали, что геморрагический шок не приводит к последующей дыхательной недостаточности, если в клиническом течении не участвовали другие факторы, связанные не с самой кровопотерей, а с ее последствиями и лечением геморрагии (например, с развитием вторичной инфекции, массивной травмой, трансфузионными реакциями, гемолизом и т. д.).
Легочное кровообращение. В настоящее время известно, что недостаточная легочная перфузия не вызывает существенных структурных повреждений легких, приводящих к СДРВ. Вместе с тем снижение перфузионного давления в сосудах легких и снижение объема легочного кровообращения приводит к функциональным расстройствам а легких. Существенно меняется соотношение вентиляция/перфузия: возникает патологический легочный шунт, являющийся главной причиной гипоксемии, увеличивается объем мертвого пространства [West J., 1974]. Этому способствует также возникающая при критических состояниях внутрисосудистая коагуляция крови (ДВС-синдром).
Инфузионная терапия и гемотрансфузия. В критическом состоянии больные, как правило, подвергаются массивной терапии в виде инфузий крови, плазмы, растворов коллоидов, кристаллоидов и других плазмозаменителей. Известно, что даже при строгом режиме приготовления официнальных растворов в них остаются мельчайшие частицы, которые при переливании задерживаются прежде всего легочными капиллярами; закупоривая их. Целлюлоза, микроскопические частицы резины, микроскопические осколки стекла, нерастворимые химические соединения, грибы и другие тела являются основным материальным субстратом, который легкие задерживают, выполняя роль биологического фильтра. До 40% переливаемых без специальных микрофильтров растворов содержат такие субстраты. В результате в легких образуются воспалительные микроскопические инфильтраты, являющиеся центрами внутрисосудистой гемокоагуляции, превращающиеся затем в воспалительные (асептические или даже септические) гранулемы. Это способствует формированию легочного патологического шунта. Целлюлоза является главным материалом подобной эмболии [Johnas H., 1967]. Она вызывает наиболее выраженную реакцию окружающих тканей. Однако легочную патологию обусловливает не только воспалительный процесс: множественные мелкие эмболы, закупоривающие мелкие капиляры легких, сами по себе приводят к повышению альвеолярного мертвого пространства, а затем и к закономерному увеличению венозного примешивания.
Обычно растворы для внутривенных инфузий на глаз вполне доброкачественны. Частицы размером меньше 40 мкм невооруженным глазом не видны даже в проходящем солнечном свете. Между тем именно такого размера частицы наиболее интенсивно задерживаются в капиллярной сети легких, вызывая патологическую картину. Очевидно, что использование специальных фильтров при любых внутривенных инфузиях, особенно массивных инфузиях у больных в критическом состоянии, крайне необходимо.
Среди множества факторов, способствующих возникновению и прогрессированию СДРВ при критических состояниях, существенное место принадлежит венозной перегрузке легких переливаемой жидкостью. При вскрытии умерших не ранее чем через 1 сут после возникновения критического состояния и начала массивной инфузионной терапии обычно обнаруживают интер-стициальный отек легких — так называемые тяжелые влажные легкие [Moore F. D. et al., 1969]. H. Jenkins и соавт. (1950) назвали подобный отек легких конгестивным ателектазированием, подчеркивая тем самым, что генез нарушений, связанных с «влажным легким», обусловлен не столько повышенным содержанием жидкости в легких, сколько спадением альвеол в результате накопления жидкости в интерстициальном пространстве.
Таким образом, главной причиной интерстициального отека легких, с которого в сущности начинается СДРВ, является передозировка жидкости на первых этапах лечения, когда представляется, что в основе патологического синдрома лежит ги-поволемия. Нередко этому способствует избыточная гемотрансфузия или введение неколлоидных растворов.
Лечение с применением высокообъемной трансфузии продолжают нередко и после того, как необходимость в этом, обусловленная циркуляторной недостаточностью, уже исчезла. Чаще такая перегрузка выявляется при наличии поражения миокарда. Избежать этого опасного явления можно лишь при хорошей координации трансфузионной программы с действительными жидкостными потребностями организма. Это возможно при внимательном мониторном контроле артериального давления, ЦВД, диуреза и в ряде случаев при контроле сердечного выброса и давления заклинивания легочной артерии. Если восстановления основных показателей гемодинамики не удается достичь инфузией жидкостных растворов или гемотрансфузией или если повышение легочного капиллярного давления (давление заклинивания) не сопровождается восстановлением системного артериального давления и не улучшает взаимоотношений в периферическом и легочном кровотоке, то следует заподозрить центральную причину (например, тампонаду сердца или, что более вероятно, ишемию миокарда и левожелудочковую недостаточность). При использовании в общей системе лечения СДРВ неколлоидных растворов (например, изотонического раствора хлорида натрия, лактата натрия или глюкозы) возникает несколько существенных моментов, которые всегда следует иметь в виду. Во-первых, может развиться тяжелая степень гипопротеине-мии, которая способствует развитию интерстициального отека легких. Важно отметить, что подобная гипопротеинемия наблюдается, как правило, при вполне удовлетворительном центральном венозном давлении.
Во-вторых, левожелудочковая недостаточность, диагностированная на основании повышения давления заклинивания легочной артерии, может развиться и при нормальном правожелудочковом конечном диастолическом давлении (нормальном давлении в правом предсердии и ЦВД) и наблюдаться достаточно долго (в несколько часов или дней), т.е. пока клапаны легочной артерии остаются состоятельными и сократимость правого желудочка не страдает.
Следовательно, левожелудочковую недостаточность, угрожающую отеком легких при инфузий изотонического раствора хлорида натрия, нельзя на первых порах диагностировать, основываясь на повышении давления в правых отделах сердца, а также по показателям ЦВД.
В-третьих, если гипопротеинемия является результатом потери крови (белков) и чрезмерной инфузий кристаллоидных растворов, то явная жидкостная перегрузка, осложняющаяся отеком легких, может проявиться значительно раньше, чем возникнут изменения ЦВД. Наиболее вероятен подобный ход событий у больных с тяжелым токсикозом, приводящим к повышению капиллярной проницаемости.
Имеется несколько ориентировочных приемов, которые помогают предупредить перегрузку жидкостью и выявить ее. Прежде всего следует количественно оценить объем жидкости, введенной больному. Доза кристаллоидов, в том числе растворов электролитов, должна соответствовать рассчитанным для таких ситуаций потребностям с учетом третьего пространства, воды местных отеков, потребности возмещения кровопотери, массы тела и, наконец, предшествующего состояния гидратации. Кроме того, следует определять гематокрит и диурез. У здоровых молодых людей при устойчивой гемодинамике почасовой диурез — наиболее чувствительный показатель гидратации. Диурез выше 80—100 мл/мин надо расценивать как предупреждение о том, что внеклеточное пространство переполнено жидкостью. Однако нужно помнить, что полиурия может отражать и полиурическую фазу почечной недостаточности.
Важность ежедневных подсчетов жидкостного баланса и обоснований объемов внутривенных инфузий в предупреждении легочной недостаточности не только следует из очевидных причин зависимости между жидкостными объемами тела, но и обусловлена исключительными трудностями коррекции этих объемов у тяжело больных. Клинически значительная передозировка кристаллоидов и электролитных растворов проявляется возникновением хрипов в легких и повышением ЦВД. Ликвидировать последствия такой передозировки прекращением введения жидкостей или инъекцией диуретиков, а также кардиотониче-ских средств удается не всегда. Массивная диуретическая терапия, например фуросемидом, может осложняться сердечными, аритмиями, особенно у больных старческого возраста, у которых раньше применялась терапия дигиталисом. Возникает неудовлетворение от такой терапии, и, главное, не устраняется, а возрастает опасность развития легочных осложнений вплоть до тяжелого отека легких, пневмонии и ОДН.
Возникновение и развитие СДРВ у больных в критическом состоянии может быть обусловлено и переливанием крови, которое в подобных случаях бывает многократным и массивным. Нередко объем перелитой крови составляет несколько литров. Ряд феноменов и риск массивных гемотрансфузий мы обсуждаем в главе «Синдромы шока и полиорганная недостаточность» и в других наших сообщениях [Рябов Г. А., 1988]. Следует подчеркнуть, что в практической деятельности реаниматолога встречаются ситуации, когда массивная гемотрансфузия при полном понимании опасностей, связанных с ней, остается главным методом лечения, способным сохранить жизнь больного.
У больных в критическом состоянии с СДРВ отрицательный эффект гемотрансфузий обусловлен в основном оседанием в капиллярах легких агрегатов эритроцитов и тромбоцитов и высвобождением из тромбоцитов вазоактивных веществ. Развивается тромбоэмболия, являющаяся основой формирования дополнительных внутрисосудистых тромбов, усиливает выраженность легочного шунта. Значение этого фактора подчеркивается тем обстоятельством, что в переливаемой донорской крови 30%, эритроцитов могут быть агрегированными. Если к этому добавить, что 25—30% перелитых эритроцитов почти немедленно секвестрируется и депонируется в различных органах и тканях, то становится очевидной необходимость всякий раз возможные выгоды гемотрансфузий соотносить с ее отрицательными результатами и искать альтернативные решения проблемы.
Мы уже указывали [Рябов Г. А., 1979] на эффект легочной и системной вазоконстрикции при гемотрансфузий. Он выражается в повышении артериального давления в обоих кругах кровообращения, обусловленном высвобождением серотонина и других вазоактивных веществ. Этот эффект не исчезает и при использовании микрофильтров, поскольку образующиеся на них пластинчатые тромбы также способны выделять вазоактивные субстраты.
Следует также упомянуть о скрытой опасности переливания свежей крови (не подвергавшейся замораживанию и малых сроков хранения). Такая опасность связана с иммунной активностью лейкоцитов, оседающих в капиллярах легких и вызывающих развитие воспалительных гранулем в легких. Замораживание крови, как известно, подавляет ее иммунную активность. Этот вопрос был хорошо изучен в экспериментах R. Nahas и соавт. (1965), S. Согг и L. Webb (1968) и др.
Внутри сосудистая коагуляция крови. Феномен свертывания крови внутри сосудов, в том числе легочных, у больных с кровопотерей и в состоянии шока впервые подробно был описан R. M. Hardaway и соавт. (1965, 1966). Активация коагуляционной системы организма приводит к образованию неустойчивых внутрисосудистых тромбов. Этот процесс находится в тесной зависимости от внутрисосудистой агрегации эритроцитов и тромбоцитов, которые при шоковой активации плазменных факторов становятся центрами образующихся тромбов. Этому способствуют капилляростаз и замедление кровотока в некоторой части легочных сосудов. Подобные мелкие тромбы могут лизироваться и исчезнуть или претерпеть процесс организации тромба.
Внутрисосудистая агрегация форменных элементов крови хорошо изучена и советскими авторами [Чернух А. М. и др., 1975]. Главное следствие подобных тромботических процессов в легких — патологическое шунтирование крови и гипоксия. В образовавшихся микросгустках крови тромбоциты высвобождают факторы, которые вызывают затем бронхоконстрикцию во всех зонах легких, что ведет к углублению так называемого синдрома промахивания и способствует разобщению вентиляции и кровотока [West J., 1974]. Аналогичные процессы происходят в тромбах, образующихся на основе агрегатов клеток при многократных переливаниях донорской крови.
Жировая эмболия. Хорошо известна жировая эмболия ветвей легочной артерии, возникающая при травматических повреждениях и, особенно, при переломах костей.
Классическая эмболия жировыми каплями вызывает тяжелые изменения в легких. У больных возникают одышка, тахикардия, затем к общим симптомам присоединяются более или менее выраженные гипоксия и гиперкапния. При рентгенологическом исследовании характерных проявлений обнаружить не удается, как и при материальной эмболии любыми другими частицами. При патологоанатомическом исследовании в зонах эмболии выявляются инфильтраты различных размеров.
В критическом состоянии возможен и другой механизм эмболии мелких ветвей легочной артерии. Он развивается в результате образования в крови нейтрального жира из свободных жирных кислот как выражение стрессовых реакций. С. Britke и соавт. (1965) назвали это явление «мобилизация жира», или «биохимическая жировая эмболизация», и связали его с длительным повышением уровня катехоламинов в крови. Содержание общих жирных кислот в сыворотке крови у больных в критическом состоянии обычно высокое и достигает иногда 6—8 г/л. Это сочетается с повышением содержания нейтральных жиров в сыворотке крови до 3—5 г/л.
Жирные кислоты в плазме крови обычно связаны с альбуминами и нетоксичны. В норме в этом состоянии они транспортируются в печень, скелетные мышцы и сердце. Появляющиеся в крови в условиях стресса и гипоксии частицы нейтрального жира под влиянием клеточных липаз постепенно гидролизуются до жирных кислот. Не связанные с белками жирные кислоты в ряде случаев могут проявлять токсические свойства. Наиболее токсична олеиновая кислота: она повреждает легочный капиллярный эндотелий, ингибирует продукцию сурфактанта, что способствует микроателектазированию в легких.
Другой механизм жировой эмболии обусловлен естественной эволюцией тромбоцитарных тромбов. Тромбоциты и образовавшиеся в сосудах белые тромбы позже распадаются и превращаются в глобулы, которые и являются частично источником жирных кислот.
Очищение легких от триглицеридов и жирных кислот можно активировать введением гепарина. Известно, что при критическом состоянии образуется дефицит эндогенного гепарина, который может быть связан как с угнетением функции печени, так и с усиленным расходом его в периферических тканях в процессе гидролиза местных жирных кислот [Goran A., Nesbakkebn R, 1969].
Токсичность кислорода. Впервые токсическое влияние кислорода на легкие было описано L. Smith в 1897 и 1899гг. Автор сделал два важных наблюдения: 1) при давлении выше, чем в обычном воздухе, кислород действует раздражающе на легкие и вызывает воспаление; 2) если легкие повреждены, то давление, при котором проявляется токсический эффект, значительно ниже того, которое требуется для здоровых легких. Таким образом, автор предупредил, что возможная токсичность О2 может ограничить его клиническое применение.
Теперь хорошо известно, что дыхание газовой смесью с высоким содержанием О2 может вызывать повреждение легких [Caldwell P. et al., 1966]. Степень повреждающего воздействия строго зависит от Ро2 вдыхаемой смеси, т. е. от F'o2 и от абсолютного давления, при котором осуществляется дыхание. Устойчивость легких к чистому О2 при низком окружающем давлении позволила использовать это явление в американских космических кораблях, где астронавты в течение нескольких недель дышат 100% О2 при давлении 0,3 атм. Возможность безопасного длительного дыхания 100% О2 была экспериментально доказана Н. Spenser (1966).
Влияние различных концентраций О2 на организм человека было изучено также в условиях повышенного атмосферного давления [Жиронкин А. Г., 1972; Петровский Б. В., Ефуни С. П., 1976; Winter S., Smith J., 1972, и др.].
С учетом широкого использования О2 в практике анестезиологии и реаниматологии представляется важным остановиться на самом главном аспекте воздействия О2 на организм — на влиянии высокого Ро2 на легкие. Практические врачи мало информированы об этой стороне кислородной терапии. В эксперименте при Ро2 дыхательной смеси выше 350—400 мм рт. ст. повреждения легких развиваются через 2—6 дней [Spenser П., 1966]. У собак возникали уплотнения легочной ткани, отек легких, кровоизлияния в них, определяемые гистологически.
Неблагоприятное влияние чистого кислорода на легкие, которое обычно развивалось в течение нескольких часов после начала дыхания 100%, О2, было подтверждено и при обследовании добровольцев [Caldwell P. et al., 1966].
Особенно выраженными оказались последствия продолжительного использования 100% О2 в сочетании с ИВЛ [Nash J., 1967]. При вскрытии умерших обнаруживали альвеолярный и интерстициальный отек легких, гистологически — расширение легочных альвеолярных перегородок, гипертрофию выстилающих клеток и гиалиновые мембраны в альвеолах^ Наиболее часто повреждения легких возникали у больных, перенесших тяжелые травмы.
В дополнение к перечисленным изменениям другие авторы указывают на истончение альвеолярных и септальных стенок, деструкцию эндотелия, некроз мембранных пневмоцитов (клетки типа I), уменьшение размеров гранулярных пневмоцитов (тип II) с последующей прогрессирующей клеточной пролиферацией [Bowden H. et al., 1968; Kapanci M. et al., 1969, и др.].
Клинические симптомы поражения паренхимы легких, возникающего под влиянием высоких концентраций О2, отражают прежде всего увеличение легочного артериовенозного шунтирования [Shapiro В. et al., 1980; Oliven A. et al., 1980] при слабо выраженном ателектазировании. Таким образом, развивающаяся у больного при дыхании 100% О2 прогрессирующая гипоксемия не может быть объяснена альвеолярным коллапсом.
В настоящее время в клинической медицине для лечения гипоксических состояний, а также для ИВЛ во время наркоза и длительной ИВЛ используют О2 только в 30—50% концентрации. Ро2 такой смеси составляет примерно 250—400 мм рт. ст., чего вполне достаточно для оксигенации крови в здоровых неповрежденных легких. По ряду причин в практических условиях иногда трудно соблюсти заданную безопасную концентрацию О2 во вдыхаемой смеси.
Аспирация. Вариант аспирации кислого желудочного содержимого (не пищи) описан С. L. Mendelson в 1946 г. и вошел в литературу под названиями «синдром Мендельсона», «острый экссудативный пневмонит» и «аспирационный синдром». Развивается острая воспалительно-экссудативная реакция слизистой оболочки бронхиального дерева с последующей обструкцией бронхиол, а также с острым воспалительным процессом в паренхиме легких. Повреждение легких возникает главным образом в тех случаях, когда рН кислого желудочного содержимого ниже 2,5. Ателектазирование массивных зон легких приводит к гиповентиляции их, нарушению соотношения вентиляция/перфузия и развитию гипоксемии.
Больные с эндотрахеальной трубкой не гарантированы от этого осложнения, так как желудочное содержимое может проникать между трубкой и трахеей, особенно при неудачном расположении трубки и при спущенной манжете. Лучший способ избежать аспирации — установить постоянный назогастральный зонд. Это должно быть обязательным и при лечении в послеоперационном периоде.
Ни при каких обстоятельствах больной не должен получить через рот значительных количеств жидкости до тех пор, пока не восстановлена удовлетворительная перистальтика. Однако, несмотря на неослабное внимание к деталям назогастральной аспирации, у некоторых больных попадание желудочного содержимого в легкие в больших или меньших количествах пока неизбежно. Это случается главным образом у больных с тяжелой черепно-мозговой травмой, а также травмой глотки и гортани, у больных, находящихся в бессознательном состянии любого происхождения, а также у больных с моторным и сенсорным параличом глотки.
При развившемся ожоге бронхов кислым содержимым желудка промывание их изотоническим раствором хлорида натрия или раствором гидрокарбоната натрия, а также последующее введение стероидных гормонов, как правило, малоэффективно.
Инфекция. В большинстве случаев бактериальная инфекция начинает играть существенную роль в патогенезе заболевания лишь в конечных стадиях синдрома. Возможно заражение следующими путями: 1) из первичного очага инфекции (перитонит, панкреатит, инфицированная рана и др.); 2) собственной кишечной флорой, ставшей для больного высоковирулентной и патогенной; 3) из окружающей среды (госпитальная инфекция); 4) из трахеостомической раны.
Развиваются различные варианты пневмонии и бронхита с обильным отделяемым, множественные септические легочные эмболические очаги, которые нередко трансформируются в абсцессы легких. Инфекция может проникать в легкие непосредственно через дыхательные пути (а также через трахеостому) или гематогенно.
В первом случае посевы содержимого бронхов свидетельствуют о совпадении инфекции с микрофлорой, характерной для данного отделения или больницы. Как правило, инфекция заносится при многократных аспирациях катетером содержимого из трахеи и бронхов, а также при длительном использовании эндотрахеальной трубки. Госпитальная инфекция наиболее интенсивно развивается у ослабленных больных. Этому способствуют отечность интерстиция легочной ткани, задержка слизи в просвете бронхов, накопление жидкости в их слизистой оболочке, нарушение дренажной функции бронхов.
Все трахеостомические раны становятся инфицированными через 2—3 сут. Обычно возбудителем служит Proteus или Pseudomonas. Фактором, предрасполагающим к развитию инфекции, является повреждение легких. Здоровые легкие, как правило, достаточно успешно противостоят трахеостомической инфекции. Нижние дыхательные пути у людей обычно стерильны. Но если дыхательные пути однажды были инфицированы, они не могут стать стерильными, пока не произойдет восстановление их анатомической целости, а также функции цилиарного аппарата бронхиол и бронхов.
Гематогенное инфицирование легких наиболее вероятно в тех ситуациях, когда имеется конкретный источник, например инфицированная рана, абсцесс или перитонит. Развивается перемежающаяся гектическая лихорадка, быстро дает рост посев крови. В легких через несколько дней находят те же микроорганизмы, что и в первичном источнике повреждения или в крови.
Преимущественное и быстрое поражение легких при наличии септического фокуса в виде абсцессов или перитонита объясняется тем, что генерализация процесса в организме происходит с обязательным участием легких как главного механического и биологического барьера для микроорганизмов.
Больные, перенесшие тяжелый шок и находящиеся в критическом состоянии, в течение нескольких дней подвергаются опасности заражения легких инфекцией из собственного желудочно-кишечного тракта. Наличие микроорганизмов в глотке, верхних дыхательных путях и кишечнике — нормальное для здорового человека явление. Как правило, подобная инфекция невирулентна. Однако она становится высоковирулентной в критическом состоянии, когда в силу тех или иных причин попадает в ткань легких и в кровеносное русло. Флора приобретает особую вирулентность через несколько дней после наложения трахеостомы. Подобные больные становятся также чрезвычайно восприимчивы к госпитальной инфекции, которая для здорового человека обычно не опасна.
Длительное применение антибиотиков направленного действия без учета чувствительности флоры больного иногда не только не способно приостановить инфекционный легочный процесс, но и повышает активность бактерий, находящихся вне сферы действия антибиотика. Особенно опасно раннее проникновение в легкие колибациллярной инфекции, в результате которого течение заболевания приобретает молниеносный характер и нередко больной погибает еще до развития картины ОДН. Разумная сдержанность в применении антибиотиков чрезвычайно важна при лечении легочных инфекционных наслоений СДРВ. Антибиотики следует выбирать в соответствии с той микробной флорой, которая дала рост в посеве содержимого из трахеи, полученного при отсасывании. Целесообразно поискать тот микроорганизм, который будет преобладать над всеми остальными в культуре. Такая ситуация характерна для больных с множественными инфицированными зонами, в частности при ожогах, когда посевы из других мест практически стерильны.
Клинический опыт показывает, что частота легочных осложнений у больных с благоприятным течением заболевания резко возрастает, если в отделение реанимации поступают больные с устойчивым инфекционным процессом (перитонит, кишечный свищ, панкреатит и др.). В таких случаях надежная изоляция подобных больных, тщательная дезинфекция отделения и аппаратуры являются единственными профилактическими мерами.
Стафилококковая инфекция является самостоятельной инфекционной проблемой у рассматриваемой группы больных. Бактериологически она выявляется довольно рано, клиническое же ее проявление зависит от степени поражения легких. Обычно это стафилококковая пневмония, выявляющаяся через несколько дней после начала заболевания, когда в организме больного находится уже несколько популяций бактерий.
В более поздних стадиях заболевания может высеваться и грибковая флора. В начале болезни она не имеет самостоятельного значения. Обычно высевается Candida albicans из крови, содержимого полых органов, мокроты и кала. Если применялись антибиотики широкого спектра, особенно в сочетании с глюкокортикоидами, то риск серьезных грибковых поражений значительно повышается. В норме грибковая флора находится в конкурентных отношениях с флорой кишечника, особенно колибациллярной. Подавление нормальной кишечной бактериальной флоры длительным применением антибиотиков является предпосылкой к усиленному росту грибков в организме, в том числе в легких.
Патофизиологические механизмы. Конечной целью лечения больного с СДРВ являются ликвидация гипоксии, восстановление функции аппарата внешнего дыхания и других органов, восстановление кислородного баланса в организме. Достижение этой цели зависит не только от качества лечения, но и от степени поражения организма, поэтому прогноз при СДРВ всегда сомнителен и неоднозначен. Между тем правильное представление о динамике развития и возможном исходе заболевания у больного с СДРВ чрезвычайно важно, так как ориентирует врача на правильные и необходимые действия.
С клинических позиций важно подчеркнуть, что при прогрессирующем СДРВ с применением интенсивной терапии, включающей ИВЛ, в большинстве -случаев опаздывают. ИВЛ начинают большей частью в третьей фазе, когда гипоксия в полном ходу и успела поразить (иногда необратимо) другие органы — мозг, почки, печень. Если пытаться оценивать в этих ситуациях субъективный фактор, то надо честно сказать, что в таком неблагоприятном ходе событий (помимо самого заболевания) чаще виновата физиологическая и реаниматологическая неосведомленность многих наших коллег — хирургов и терапевтов, искренне убежденных, что если больного «сажают» на аппарат, то «ему конец». Между тем предупредить губительное действие гипоксии еще во второй фазе СДРВ — одна из главнейших задач лечения. К сожалению, это не всегда удается, даже если все сделано вовремя.
Именно поэтому так важно оценить прогноз заболевания. Он бывает более определенным, когда удается в наиболее ранние сроки установить характер самых незначительных патофизиологических сдвигов, дать научно обоснованную трактовку их. Мы убеждены, что врач может предвидеть наиболее вероятное течение патологического процесса и на этой основе своевременно применить необходимые лечебные меры. В многообразном и запутанном СДРВ трудно разобраться даже опытному реаниматологу. Условия лечения подобных больных требуют не просто замены нарушенной функции (в данном случае функции дыхания), но также глубинного понимания патофизиологии происходящих расстройств, позволяющего правильно применить адекватное интенсивное общее лечение.
Диффузия газов и альвеолярно-капиллярная блокада. Переход газов из альвеол в крови и обратно представляет собой диффузию газов через проницаемую для них мембрану: молекулы газа переходят из зоны высокого в зону низкого парциального давления. Следовательно, у человека диффузия может быть выражена объемом газов (например, О2)г который способен пройти через альвеолярно-капиллярную мембрану за 1 мин при градиенте парциальных давлений, равном 1 мм рт. ст. У здорового человека диффузионная способность легких для О2 составляет 15—20 мл/(мин · мм рт. ст.). Эта величина возрастает при физической нагрузке. Через всю поверхность здоровых легких в организм может проникнуть более 6 л О2 в минуту. Обычно в клинической практике эту величину не определяют, поскольку результаты, полученные при ее расчете, сугубо ориентировочны.
J. Austrian и соавт. (1951) назвали нарушение диффузионной способности легких альвеолярно-капиллярной блокадой. Они обратили внимание на то, что синдром альвеолярно-капиллярной блокады неспецифичен. Он возникает при тяжелых заболеваниях и поражениях легких. Патогенез этого явления сложен и до конца не изучен. Во всяком случае, стало очевидным, что синдром альвеолярно-капиллярной блокады может быть обусловлен многочисленными причинами. В пользу этого говорит сложность структуры альвеолярно-капиллярной мембраны, обусловливающая многоступенчатость самого процесса переноса газов. Помимо основного фактора простой физической диффузии, в процессе переноса газов через мембрану участвуют и факторы, активирующие его и связанные с наличием в альвеолярно-капиллярной мембране особых образований — везикул, ускоряющих активный перенос веществ через мембрану, а также с наличием белков и липидов особой структуры, выстилающих поверхность алвеолы.
Компоненты патологии легочной ткани при СДРВ, которые определяют нарушение диффузии газов через альвеолярно-капиллярную мембрану, хорошо известны, хотя роль каждого из них мало изучена. Наиболее значимыми из этих компонентов являются интерстициальный (перикапиллярный) отек, гипертрофия альвеолярных клеток, образование гиалиновых мембран внутри альвеол и интерстициальный фиброз легких. С клинических позиций все четыре компонента следует рассматривать вместе как общую причину возникающего увеличения дистанции прохождения молекул О2 и СО2 и увеличения барьера между пространством альвеолы и эритроцитарным гемоглобином. В конечном счете парциальное давление газов в крови зависит от «дистанции пробега» и от разности парциальных давлений газа по обе стороны мембраны. С физиологической точки зрения в нормальных легких диффузия О2 через альвеолярно-ка-пиллярную мембрану имеет широкие пределы. Если у больного с увеличенной «дистанцией пробега», вызванной образованием тканевого легочного барьера, применять 100% О2, т. е. увеличить разность парциальных давлений O2 между альвеолярным газом и кровью, то можно снизить «диффузионные потери». Однако больной должен долго дышать 100% О2, чтобы достичь полного вымывания N2 из образовавшихся альвеолярных ловушек. Практически в начальных фазах СДРВ для этого требуется не менее 30—45 мин.
Синдром капиллярного просачивания и отек легких. Развитие интерстициального и альвеолярного отека с повышением левопредсердного или легочного венозного давления является следствием закона Старлинга, определяющего условия транскапиллярного жидкостного обмена. Механизмы, ответственные за развитие отека легких при нормальном легочном венозном давлении (так называемого отека с низким легочным давлением), достаточно сложны. Обычно называют три причины, обусловливающие отек: снижение онкотического давления плазмы, повышение легочной капиллярной проницаемости, изменение функции легочных лимфатических сосудов [Sta-ub N. С., 1974]. Первые два механизма часто комбинируются и приводят к увеличению скорости лимфообращения и повышению концентрации белков в лимфе легких [Demling R. H. et al., 1979]. Роль лимфы в поддержании нормальной анатомии и функции интерстициального легочного пространства исключительно велика. Однако массивные инфузии жидкостей, развитие инфекции, введение лекарств и изменения внутрилегочного давления существенно влияют на лимфатическую систему легких и могут способствовать возникновению отека их~. Если возникает функциональная блокада тока лимфы в сочетании с выраженным повышением внутрибронхиального давления, то в неподатливых легких жидкость быстро накапливается в легочном ин-терстиции даже при относительно малом изменении онкотического давления или легочной мембранной проницаемости.
ОДН, которая развивается в результате синдрома капиллярного просачивания и отёка легких, может быть двух типов. ОДН первого типа характеризуется развитием так называемого влажного легкого и оценивается сейчас как более благоприятное состояние. На фоне интерстициального отека легких определяется нормальное легочное капиллярное давление (судят по результатам исследования давления заклинивания легочной артерии) и отсутствует легочная гипертензия. Диуретическая терапия с использованием фуросемида, ультрагемофильтрация или ограничение жидкостной нагрузки достаточно эффективны.
При ОДН второго типа течение болезни более тяжелое, прогноз неблагоприятен. Главным патологическим симптомом является легочная гипертензия на фоне интерстициального отека легких. Легочная ангиография не выявляет легочных капилляров, которые при этом варианте бывают заполнены фибрино-выми (иногда эритроцитарными) микроэмболами. Обычно подобное состояние сочетается с выраженным ДВС-синдромом, преимущественно с его первой фазой (гиперкоагуляции). ОДН развивается стремительно и характеризуется тяжелой гипоксе-мией, обусловливающей необратимость заболевания. При этом, помимо мероприятий по поддержанию адекватной оксигенации крови (ИВЛ или как крайней меры — экстракорпоральной мембранной оксигенации), показано лечение с использованием стрептокиназы и гепарина.
Патология перфузии легких. Нарушения вентиляции при СДРВ происходят одновременно с расстройствами кровообращения в легких. Наиболее отчетливые нарушения легочного кровообращения развиваются главным образом в венозной системе и выражаются преимущественно в тромбоэмболии; при этом кровь механически шунтируется в неповрежденные сосудистые зоны. Тромбоциты в сгустках крови начинают высвобождать факторы, которые вызывают бронхоконстрикцию во всех зонах легких, что ведет к углублению синдрома «промахивания» и разобщению вентиляции и перфузии. Те же процессы, хотя и менее выраженные, наблюдаются в эмболах, возникающих в результате многократных переливаний крови. Наиболее общей причиной перераспределения легочного кровотока является левожелудочковая недостаточность с повышением давления в левом предсердии. При этом повышенное легочное венозное давление способствует увеличению легочного кровотока в области плохо вентилируемых зон легких и таким образом увеличивает шунтирование. Увеличение количества внесосуди-стой жидкости в легких, вызванное повышением легочного капиллярного давления, содействует закрытию малых дыхательных путей и коллапсу альвеол.
Другие причины увеличения легочного шунтирования при СДРВ включают механизмы, важность которых несомненна, но которые с клинических позиций бывает трудно оценить. Первый из них связан с ускорением пассажа эритроцита через легочные капилляры. Как известно, у большого числа больных на первых этапах развития СДРВ наблюдается гипердинамический синдром, который характеризуется очень высоким сердечным выбросом. Обычно регистрируют увеличение сердечного выброса в 2—3 раза в ранних стадиях после начала лечения. Особенно выражен этот синдром у больных, состояние которых осложнено сепсисом. С позиций физиологии сердечный выброс должен быть приспособлен по объему к уменьшенному числу легочных капилляров. В результате продолжительность среднего транзитного времени для каждого, эритроцита существенно снижается. Очевидно, что при таких обстоятельствах, особенно если усилен диффузионный барьер или имеется гиповентиляция, развивается неполная оксигенация гемоглобина, что может восприниматься как увеличение легочного шунтирования [West J. В., 1974]. Этот эффект еще более усиливается, если кровь, проникающая в легкие, имеет ацидотическую реакцию, которая смещает кривую оксигенации гемоглобина вправо.
Гипервентиляция и гипокапния. В реаниматологической практике высокий минутный объем дыхания (MOB) называют «гипервентиляцией». Этим термином можно охарактеризовать не только спонтанное дыхание больного, но также режим ИВЛ. С точки зрения физиологии гипервентиляция (одышка) представляет собой усиленный режим дыхания, обусловленный активацией дыхательного центра под влиянием изменившихся условий внутренней среды в организме, в частности при снижении РаО2 при возникновении ацидоза или повышении температуры тела.
Нарушения ритма и интенсивности дыхания являются также нормальной реакцией на тяжелую травму, боль, страх, раздражение брюшины. С учетом этого, по-видимому, более правильно не столько искать внутреннюю причинную связь между возникающей гипервентиляцией и биохимическими изменениями внутренней среды (хотя это тоже необходимо), сколько принять к сведению факт гипервентиляции как первого симптома развивающейся дыхательной недостаточности.
Ранний период развития дыхательных расстройств у больных в критическом состоянии характеризуется гипервентиляцией, приводящей к гипокапнии и умеренному респираторному алкалозу. Подобная спонтанная гипервентиляция может, по-видимому, иметь отношение к этиологическому фактору, т. е. может быть вызвана травмой, кровопотерей или инфекционным фактором лишь в самом начале.
Не существует убедительного физиологического объяснения раннего появления гипервентиляции. Однако известно, что в одних случаях это происходит на фоне ранней неадекватной оксигенации или позже, при полном развитии СДРВ, когда гипервентиляция может быть следствием развивающейся гипоксии, и тогда ее появление кажется вполне закономерным. В других случаях (пожалуй, в большинстве) гипервентиляция появляется без признаков гипоксии. Выраженная, гипокапния и обусловленный ею алкалоз нежелательны всегда, особенно у больных с пороками сердца, периферической сосудистой недостаточностью, аритмией сердца или при лечении дигиталисом. Поскольку гипокапния и дыхательный алкалоз — непременное следствие и аппаратной гипервентиляции, целесообразно по возможности предупреждать развитие этого состояния увеличением объема мертвого пространства с помощью дополнительной вставки между эндотрахеальной трубкой и респиратором. Алкалоз плохо переносится человеком. Его кажущаяся безвредность в противоположность ацидозу частично является результатом логарифмической природы рН-шкалы: равные изменения рН при алкалозе сопровождаются меньшими изменениями [Н+], чем при ацидозе.
Респираторный алкалоз оказывает существенное влияние на деятельность мозга. Этот вопрос был изучен J. Hinshaw и R. Booth (1941) при оценке деятельности пилотов в реальных условиях. При исследовании реакции пилотов в стрессовых ситуациях авторы обнаружили, что гипервентиляция нарушает точность различных производимых пилотами привычных процедур и объясняли это церебральной ишемией. S. S. Kety и С. F. Schmidt (1946) в хорошо известных теперь исследованиях, определяя изменения церебрального кровотока под влиянием изменений Рсо2, установили, что при спонтанной гипервентиляции у добровольцев, приводившей к снижению Рсо2 до 18 мм рт. ст., мозговой кровоток уменьшался на 32%. Продолжением этих исследований явились работы J. S. Meyer и F. Gotoh (1966), в которых в экспериментах на кошках было показано, что гипервентиляция, снижавшая Рсо2 до 10 мм рт. ст., существенно снижала Ро2 в коре мозга. При этом возникали также гистологические изменения в мозге.
Другие эффекты, вызванные респираторным алкалозом, выражаются в вазоконстрикции, снижении артериального давления, повышении числа циркулирующих эритроцитов при снижении общего объема циркулирующей плазмы, наконец, снижением концентрации Са2+ при повышении содержания общего кальция плазмы [Robinson J. S., Gray T. С., 1961].
Расстройства периферического кровообращения и лактат-ацидоз. Уже на ранних стадиях развития синдрома дыхательных расстройств наблюдается умеренное повышение концентрации L—. Обычно развивающийся лактат-ацидоз сочетается с низким сердечным выбросом, начальным дыхательным алкалозом и гипокапнией. Во второй фазе синдрома можно наблюдать нормализацию уровня L— в крови. Однако с переходом в третью и особенно в четвертую фазу вновь можно отметить повышение содержания L— в крови и лактат-ацидоз, которые развиваются теперь на фоне расстройств системного кровообращения. Подобная динамика содержания l- в крови и ацидоза характерна для больных с гиподинами-ческим синдромом. Однако описанную динамику L—крови вряд ли можно объяснить лишь системной циркуляторной недостаточностью и самим СДРВ, обусловливающими гипоксию. По-видимому, здесь действуют более сложные и неоднозначные факторы. Повышение уровня L— в крови наблюдается, в частности, при гипервентиляционной гипокапнии и в отсутствие существенных нарушений системного кровообращения. При этом содержание L— в крови быстро нормализуется при прекращении гипервентиляции [Robinson J.S., Gray Т. С., 1961]. Описано также повышение концентрации l- в крови после введения NaHCO3 [Haldi H., 1933].
W. Huckabee (1958), а затем его последователи показали, что пропорционально повышению L~ в крови повышается и уровень пирувата.
Лактат является нормальным метаболитом глюкозы, появляющимся в результате тканевого анаэробного гликолиза. При образовании L—появляется эквимолярное количество Н+. Это и определяет возможный сдвиг КОС в сторону ацидоза. Образование L— не дает организму существенной энергетической прибыли: большая часть калорического выхода при метаболизме глюкозы получается путем окислительного фосфорилирования.
Повышение уровня L— в первой фазе СДРВ бывает обычно умеренным (до 2—3 ммоль/л) и наблюдается 2—3 дня. Если кровообращение при этом стабилизируется, то большая часть L— может метаболизироваться и уровень его постепенно приходит к норме. При прогрессировании СДРВ с увеличением венозного примешивания и углублением артериальной гипоксе-мии как при гиподинамии, так и при гипердинамии гипоперфузия тканей усиливается. Концентрация L—продолжает повышаться и в ряде случаев достигает 4—6 ммоль/л. Обычно это сопровождается высоким отношением лактат/пируват.
Таким образом, системная гипоперфузия — наиболее важная причина лактацидемии. Другая очевидная причина лактат-ацидоза — артериальная гипоксемия — при тех величинах РаO2, которые допускают жизнеспособность тканей, как свидетельствуют данные W. Huckabee (1958), в общем не ведет к избытку продукции лактата. Все же лактацидемия является достаточно надежным индикатором гипоксии организма. При этом гипоксию следует рассматривать как глобальное явление для организма, возникающее в результате не одного, а множества факторов.
Возможность охарактеризовать гипоксию по концентрации лактата крови была использована W. Huckabee (1958). Автор взял за основу уравнение восстановления пирувата в лактат при участии систем NAD/NADH и вывел формулу, позволившую вычислить тот «избыточный» лактат, появление которого не объясняется исходной концентрацией лактата и пирувата. Этот показатель обозначает как ExL. Концепция W. Huckabee широко распространилась и используется в научных изысканиях по проблеме гипоксии.'По мнению этого автора и других исследователей, показателей ExL наилучшим образом коррелирует с другими показателями, характеризующими гипоксию, и с клиническими данными.
Однако концепция W. Huckabee вызвала также и критику. Основные аргументы возражающих сводятся к указаниям на отсутствие равновесных состояний реакций в живом организме, на различное содержание лактата и пирувата внутри и вне клетки, а также на изменения константы диссоциации. При сниженной перфузии тканей в кровеносное русло попадает большое количество L—, особенно из скелетных мышц. В норме печень и миокард способны к самоочищению от L—, т. е. могут активно метаболизировать его. При расстройствах периферического кровообращения, малом сердечном выбросе и в условиях гипоксемии это становится невозможным. При концентрации L,- в крови свыше 10 ммоль/л больные не выздоравливают. При этом почти весь лактат (свыше 80% его) определяется в виде ExL. В отсутствие почечной недостаточности или диабетического ацидоза некоторое снижение BE может объясняться, в частности, и повышением концентрации l- в крови. Таким образом, уровень лактата в крови можно считать прогностическим кри

