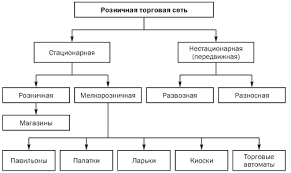
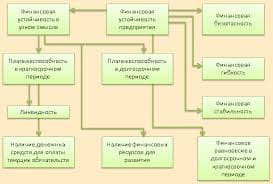
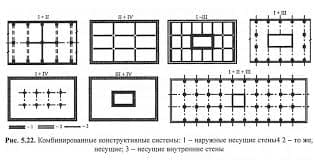
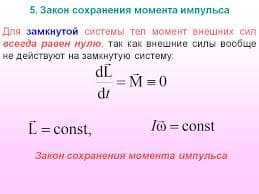2015-07-14
2015-07-14 558
558Местные анестетики
Антиаритмические средства
Фармакологическое действие - местноанестезирующее, антиаритмическое.
Антиаритмическая активность обусловлена угнетением фазы 4 (диастолической деполяризации) в волокнах Пуркинье, уменьшением автоматизма, подавлением эктопических очагов возбуждения. На скорость быстрой деполяризации (фаза 0) не влияет или незначительно снижает. Увеличивает проницаемость мембран для ионов калия, ускоряет процесс реполяризации и укорачивает потенциал действия. Не изменяет возбудимость синусно-предсердного узла, мало влияет на проводимость и сократимость миокарда. При в/в введении действует быстро и коротко (10–20 мин).
Механизм местноанестезирующего эффекта заключается в стабилизации нейрональной мембраны, снижении ее проницаемости для ионов натрия, что препятствует возникновению потенциала действия и проведению импульсов. Возможен антагонизм с ионами кальция. Быстро гидролизуется в слабощелочной среде тканей и после короткого латентного периода действует в течение 60–90 мин. При воспалении (тканевой ацидоз) анестезирующая активность снижается. Эффективен при всех видах местного обезболивания. Расширяет сосуды. Не оказывает раздражающего действия на ткани.
При в/в введении C max создается практически «на игле» (через 45–90 с), при в/м — через 5–15 мин. Достаточно быстро абсорбируется со слизистой оболочки верхних дыхательных путей или полости рта (C max достигается через 10–20 мин). После приема внутрь биодоступность составляет 15–35%, так как 70% всосавшегося препарата подвергается биотрансформации при «первом прохождении» через печень. В плазме на 50–80% связывается с белками. Стабильная концентрация в крови устанавливается через 3–4 ч при непрерывном в/в введении (у больных с острым инфарктом миокарда — через 8–10 ч). Терапевтический эффект развивается при концентрации 1,5–5 мкг/мл. Легко проходит через гистогематические барьеры, включая ГЭБ. Сначала поступает в хорошо кровоснабжающиеся ткани (сердце, легкие, мозг, печень, селезенка), затем — в жировую и мышечную ткани. Проникает через плаценту, в организме новорожденного обнаруживается 40–55% концентрации роженицы. Экскретируется в грудное молоко. T 1/2 после в/в болюсного введения — 1,5–2 ч (у новорожденных — 3 ч), при длительных в/в инфузиях — до 3 ч и более. При нарушении функции печени T 1/2 может увеличиваться в 2 раза и более. Быстро и почти полностью метаболизируется в печени (в неизмененном виде с мочой выводится менее 10%). Основной путь деградации — окислительное N-дезалкилирование, при этом образуются активные метаболиты (моноэтилглицинксилидин и глицинксилидин), имеющие T 1/2 2 ч и 10 ч соответственно. При хронической почечной недостаточности возможна кумуляция метаболитов. Длительность действия — 10–20 мин при в/в введении и 60–90 мин — при в/м.
При местном применении на неповрежденную кожу (в виде пластин) возникает терапевтический эффект, достаточный для снятия болевого синдрома, без развития системного эффекта.