 2014-05-17
2014-05-17 5598
5598Основная задача теорий химической кинетики - предложить способ расчета константы скорости элементарной реакции и ее зависимости от температуры, используя различные представления о строении реагентов и пути реакции. Существуют несколько теорий химической кинетики. Наиболее известными являются теория активных столкновений (ТАС) и теория переходного состояния (называемая также теорией активированного комплекса).
На основе молекулярно-кинетической теории газов была разработана для бимолекулярных реакций теория, получившая название теории активных соударений (ТАС). Согласно этой теории реакция осуществляется при столкновении между молекулами реагирующих веществ. Во время столкновений происходит разрыв старых и образование новых химических связей.
Рассмотрим для примера бимолекулярную реакцию, в которой превращение осуществляется в результате соударения двух молекул А и В. Упрощенный расчет числа соударений между молекулами А и В в единицу времени в единице объема, основанный на молекулярно-кинетической теории газов, дает
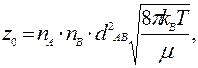
где п — число молекул в единице объема; dAB = ½(dA + dB), d - эффективный диаметр, 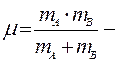 приведенная масса.
приведенная масса.
Если бы каждое соударение приводило к реакции, то скорость реакции, определяемая как изменение количества вещества в молях за единицу времени в единице объема (л), была бы равна
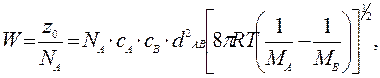
Вводя долю активных соударений и учитывая, что для бимолекулярных реакций
W = k∙cA∙cB,
Получим
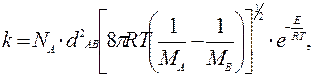
или
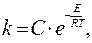
где С – константа.
Для того чтобы привести теорию в соответствие с экспериментом, вводят так называемый стерический множитель Р и выражение для константы скорости записывают в виде
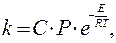
Для различных реакций Р имеет величину от 1 до 10-8. Возможной причиной отклонений является необходимость определенного расположения молекул друг относительно друга при соударениях. Кроме того, избыточная энергия может не успеть перераспределиться между связями так, чтобы произошла реакция, и молекулы разойдутся, не прореагировав. Таким образом, стеричский множитель Р = кэ/кр характеризует вероятность протекания реакции при активных соударениях.
Однако теория соударений не дает возможности вычислить величину Р. Более того, в рамках теории соударений нет объяснения тому факту, что для некоторых реакций Р > 1 (так называемые быстрые реакции).
В 1935 г. Г. Эйринг, М. Поляни и М. Эванс предложили теорию, которая принципиально дает возможность рассчитать не только стерический фактор, но и энергию активации исходя из свойств участников реакции.
Эта теория получила название теории переходного состояния, или теории активированного комплекса.
Согласно этой теории при протекании элементарной химической реакции исходные вещества переходят из основного энергетического состояния в возбужденное. Этот переход сопровождается изменением конфигурации реагирующих частиц и изменением потенциальной энергии состоящей из них системы реагирующих частиц. Например, в ходе реакции
А + ВС → АВ + С
расстояние между атомами А и В уменьшается, а между атомами В и С увеличивается. Если построить график зависимости потенциальной энергии системы (Z) от расстояний у и х между атомами А—В и В—С соответственно при фиксированной величине угла АВС, то получим некоторую поверхность, называемую поверхностью потенциальной энергии. Каждому расположению частиц А, В и С соответствует одно значение потенциальной энергии и одна точка на этой поверхности.
Наиболее выгодный (с точки зрения затрат энергии) путь перехода от исходной системы к конечной проходит по линии, соединяющей точки, соответствующие минимуму потенциальной энергии. Путь вдоль нее называют координатой реакции. Зависимость потенциальной энергии реагирующих частиц от координаты реакции имеет вид кривой с максимумом.
Промежуточное состояние системы реагирующих частиц, соответствующее этому максимуму, называется переходным состоянием или активированным комплексом. В теории активированного комплекса переходное состояние рассматривают как «обыкновенную молекулу, обладающую обычными термодинамическими свойствами, за исключением того, что движение вдоль координаты реакции приводит к распаду с определенной скоростью. Сделав это допущение, при помощи статистических методов можно найти концентрацию активированных комплексов и скорость их перехода через критическую конфигурацию активированного состояния (Г. Глесстон, К. Лейдлер, Г. Эйринг. Теория абсолютных скоростей реакций. М., 1948).
Основной постулат теории активированного комплекса состоит в том, что исходные вещества всегда находятся в равновесии с активированным комплексом. В соответствии с этим схема записанной выше реакции должна выглядеть следующим образом:
А + ВС 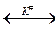 АВС
АВС 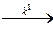 АВ + С.
АВ + С.
Здесь К# = 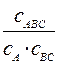 - константа равновесия реакции образования активированного комплекса.
- константа равновесия реакции образования активированного комплекса.
Скорость образования продуктов реакции
w = k'∙ савс = к'∙К#∙ сА∙ сВC
С другой стороны, если время жизни активированного комплекса обозначить через τ#, то скорость его распада, т. е., скорость интересующей нас реакции, будет равна
W= cABC/ τ#.
Методами статистической физики можно найти, что
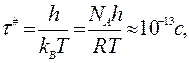
где h — постоянная Планка; kB – постоянная Больцмана.
Отсюда получим, что
W = 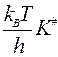 ∙сА∙ сВС,
∙сА∙ сВС,
В результате получает формулу закона действующих масс, в которой множитель перед произведением концентраций является константой скорости реакции, независящей от концентрации реагирующих веществ
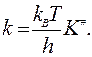
Величину К# можно рассчитать методами статистической физики через так называемые статистические суммы Qi исходных веществ и активированного комплекса:
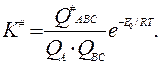
Для определения статистических сумм надо знать молекулярные массы, моменты инерции и частоты колебаний исходных молекул и активированного комплекса. Наиболее трудной задачей является расчет Q#ВС, поскольку неизвестны конфигурация и молекулярные постоянные активированного комплекса и методов их экспериментального определения пока нет.; Тем не менее разработаны приближенные методы, позволяющие провести достаточно надежную оценку Q#.
Величина E0 представляет собой разницу нулевых энергий активированного комплекса исходных молекул, т. е. соответствует энергии активации при Т = 0. Величина E0 может быть рассчитана, если известна поверхность потенциальной энергии. К сожалению, в настоящее время точный расчет поверхности потенциальной энергии возможен лишь для нескольких систем из изотопно замещенных молекул водорода. В остальных случаях используют различные полуэмпирические методы, основанные на тех или иных допущениях.
Несмотря на имеющиеся трудности, теория активированного комплекса дает принципиальную возможность теоретического расчета констант скорости реакций. Поэтому ее называют еще теорией абсолютных скоростей реакции.
Другим важным результатом теории является возможность связать константу скорости с изменением термодинамических функций реакции образования активированного комплекса.
Поскольку
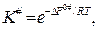
то
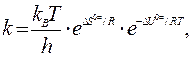
где ∆S0# и ∆U0# - стандартные изменения энтропии и внутренней энергии указанной реакции. Их называют также энтропией и энергией активации.
Сравнение последнего уравнения с уравнением теории соударений дает возможность установить, фактор соударений С равен 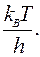 Обе величины имеют размерность частоты (время-1), и при 298 К их значения близки к 10-13 с-1.
Обе величины имеют размерность частоты (время-1), и при 298 К их значения близки к 10-13 с-1.
Энергия активации соответствует энергии образования активированного комплекса, а стерический множитель расшифровывается через изменение энтропии того же процесса как 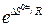 .
.
Величина ∆S0# может быть положительной или отрицательной в зависимости от того, будет ли активированный комплекс менее компактен, чем исходные реагенты, или нет.
Последнее уравнение позволяет также объяснить существование быстрых процессов с большими энергиями активации и медленных с малыми энергиями активации. Например, денатурация протеинов характеризуется очень высокой энергией активации, однако эта реакция идет быстро, так как очень сильно возрастает энтропия.

