 2015-03-22
2015-03-22 1261
1261При разработке теории строения молекул, в начале 30-х годов были выдвинуты различные приближённые квантовохимические теории, более или менее удовлетворительные в зависимости от характера рассматриваемых задач: теория валентных связей, заложенная в 1927 В. Гейтлером и Ф. Лондоном в Германии, а в начале 30-х гг. развитая Дж. Слейтером и Л. Полингом в США; теория кристаллического поля, предложенная немецким учёным Х. Бете в 1929 и в последующие годы разрабатывавшаяся американским учёным Ван - Флеком (своё применение в химии она получила в 1950-е гг. как теория поля лигандов благодаря исследованиям английского учёного Л. Оргела и датских учёных К. Йоргенсена и К. Бальхаузена). В конце 20-х гг. XX ст. были заложены основные принципы теории метода молекулярных орбиталей, разработанного Дж. Леннардом-Джонсом (Великобритания), Р. Малликеном (США), Ф. Хундом (Германия) и развивавшегося затем многими др. исследователями. Долгое время эти приближённые теории сосуществовали и даже дополняли друг друга. Однако теперь, когда достигнуты огромные успехи в синтезе молекул и определении их структуры, а вычислительная техника получила широкое развитие, симпатии исследователей склонились в сторону метода молекулярных орбиталей. Это объясняется тем, что только в рамках метода молекулярных орбиталей был выработан универсальный язык, пригодный для описания любых молекул, строение которых отличается очень большим разнообразием и сложностью. Метод молекулярных орбиталей включает наиболее общие физические представления об электронном строении молекул и, что не менее важно, использует математический аппарат, наиболее пригодный для проведения количественных расчётов на ЭВМ. В основе метода молекулярных орбиталей лежит одноэлектронное приближение, в рамках которого каждый электрон считается квазинезависимой частицей и описывается своей волновой функцией. Обычно в основе большинства вычислительных лежит метод линейных комбинаций Релея – Ритца, в рамках которого все одноэлектронные молекулярные орбитали получаются как линейные комбинации соответствующих исходных атомных орбиталей. Метод молекулярных орбиталей исходит из того, что каждый электрон молекулы находится в поле всех её атомных ядер и остальных электронов. При этом, соответствующие атомные орбитали, описывающие электронное строение атомов, включаются в метод молекулярных орбиталей как частный случай, когда в системе имеется только одно атомное ядро. Далее, в рамках данного метода рассматривают все химические связи как многоцентровые (по числу атомных ядер в молекуле) и тем самым полностью делокализованные. С этой точки зрения всякого рода преимущественная локализация электронной плотности около определённой части атомных ядер есть приближение, обоснованность которого должна быть выяснена в каждом конкретном случае. Представления В. Косселя о возникновении в химических соединениях обособленных ионов (изоэлектронных атомам благородных газов) или воззрения Дж. Льюиса (США) об образовании двухцентровых двухэлектронных химических связей (выражаемых символикой валентного штриха) естественно включаются в теорию МО как некоторые частные случаи. Таким образом, в методе молекулярных орбиталей молекула рассматривается с той же точки зрения, что и атом, т.е. принимается, что электроны в молекуле находятся на молекулярных орбиталях, охватывающих все её ядра. В отличие от атомных орбиталей, молекулярная орбиталь является многоцентровой. Молекулярной орбитали присущи все фундаментальные принципы и свойства атомных орбиталей: принцип Паули, правило Гунда, свойства симметрии и т.д. Существуют различные варианты составления молекулярных орбиталей, однако наиболее наглядным, информативным и удобным оказался метод ЛКАО - МО, в котором молекулярная орбиталь записывается как линейная комбинация всех атомных орбиталей, участвующих во взаимодействии. Волновая функция основного состояния молекулы задаётся как произведение одноэлектронных волновых функций занятых молекулярных орбиталей:
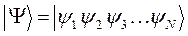
при этом для молекулы существует строго определённый набор дозволенных состояний: 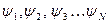 , которым отвечают определённые значения энергии. Данное представление является, конечно, приближённым, поскольку оно основано на предположении, что вблизи каждого атомного ядра молекулярная орбиталь похожа на соответствующую атомную орбиталь, тогда как на самом деле функция распределения будет более сложной. Наиболее точные расчёты в приближении метода молекулярных орбиталей выполняются методом самосогласованного поля (ССП) Хартри – Фока. Молекулярные орбитали вычисленные в рамках данного метода, наиболее близки (хотя и не всегда) к истинным и называются орбиталями Хартри – Фока или спектроскопическими. Свойства молекулярной орбитали находятся в прямой зависимости от свойств соответствующих атомных орбиталей. Набор атомных орбиталей, используемых при квантовохимическом расчёте молекул, называется атомным базисом. Существует три основных критерия для выбора базисных функций: базисные функции должны давать хорошее приближение к истинной волновой функции и должны допускать аналитическое вычисление нужных интегралов. Кроме того, полное число базисных функций не должно быть очень большим. Лучшими базисными функциями как правило являются самосогласованные, т.е. вычисленные с помощью метода ССП, атомные орбитали, найденные Клементи и Ватсоном путём решения уравнения Хартри - Фока. Однако эти функции получаются не в аналитическом виде, а в табличном. Проводить расчёты с функциями, заданными в численном виде, крайне трудно и неудобно. В качестве базиса атомных орбиталей можно использовать слэйтеровские орбитали. Форма волновой функции, предложенная Слейтером, тесно связана с видом атомных орбиталей водородоподобных атомов:
, которым отвечают определённые значения энергии. Данное представление является, конечно, приближённым, поскольку оно основано на предположении, что вблизи каждого атомного ядра молекулярная орбиталь похожа на соответствующую атомную орбиталь, тогда как на самом деле функция распределения будет более сложной. Наиболее точные расчёты в приближении метода молекулярных орбиталей выполняются методом самосогласованного поля (ССП) Хартри – Фока. Молекулярные орбитали вычисленные в рамках данного метода, наиболее близки (хотя и не всегда) к истинным и называются орбиталями Хартри – Фока или спектроскопическими. Свойства молекулярной орбитали находятся в прямой зависимости от свойств соответствующих атомных орбиталей. Набор атомных орбиталей, используемых при квантовохимическом расчёте молекул, называется атомным базисом. Существует три основных критерия для выбора базисных функций: базисные функции должны давать хорошее приближение к истинной волновой функции и должны допускать аналитическое вычисление нужных интегралов. Кроме того, полное число базисных функций не должно быть очень большим. Лучшими базисными функциями как правило являются самосогласованные, т.е. вычисленные с помощью метода ССП, атомные орбитали, найденные Клементи и Ватсоном путём решения уравнения Хартри - Фока. Однако эти функции получаются не в аналитическом виде, а в табличном. Проводить расчёты с функциями, заданными в численном виде, крайне трудно и неудобно. В качестве базиса атомных орбиталей можно использовать слэйтеровские орбитали. Форма волновой функции, предложенная Слейтером, тесно связана с видом атомных орбиталей водородоподобных атомов:
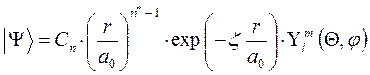
здесь:
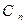 - нормировочный множитель;
- нормировочный множитель;
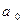 - боровский радиус атома водорода;
- боровский радиус атома водорода;
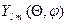 - угловая составляющая та же, что и для водородоподобных функций;
- угловая составляющая та же, что и для водородоподобных функций;
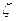 - орбитальная экспонента;
- орбитальная экспонента;
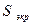 - константа экранирования, учитывающая сумму вкладов, вносимых отдельными электронами за вычетом рассматриваемого электрона;
- константа экранирования, учитывающая сумму вкладов, вносимых отдельными электронами за вычетом рассматриваемого электрона;
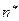 - эффективное квантовое число, совпадающее со значением главного квантового числа
- эффективное квантовое число, совпадающее со значением главного квантового числа 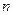 для орбиталей первых трёх периодов, а для четвёртого, пятого и шестого оно соответственно равно 3,7; 4,0 и 4,2.
для орбиталей первых трёх периодов, а для четвёртого, пятого и шестого оно соответственно равно 3,7; 4,0 и 4,2.
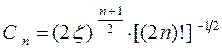
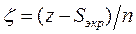
Константа экранирования расчитывается по следующим правилам:
1. Все атомные орбитали классифицируются по группам: (1s); (2s, 2p); (3s, 3p); (3d); (4s, 4p); (4d, 4f) и т.д., то есть s- и p-орбитали одной оболочки группируются вместе, а d- и f-орбитали представляют отдельные группы.
2. Константа 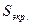 определяется как сумма следующих вкладов: 0,35 от каждого электрона в рассматриваемой группе (в случае 1s-группы – 0,3); 0,85 от каждого электрона ближайшей внутренней группы (s или p); 1,0 - от каждого электрона более глубоких слоёв (d или f). При этом нужно иметь в виду, что учитываются только электроны, находящиеся между ядром и исследуемой оболочкой. Электроны, внешние по отношению к исследуемому, в экранировании участия не принимают. При вычислении констант экранирования для данной электронной конфигурации необходимо вместо 1s, 2s, 2p и т.д. в правой части равенств подставить число электронов на данной орбитали. Радиальные функции слейтеровского типа недостаточно точно описывают поведение атомных орбиталей Хартри – Фока на небольших расстояниях от ядра. Этот недостаток можно устранить, если использовать для аппроксимации атомной орбитали Хартри – Фока по крайней мере две слейтеровские функции с разными орбитальными экспонентами:
определяется как сумма следующих вкладов: 0,35 от каждого электрона в рассматриваемой группе (в случае 1s-группы – 0,3); 0,85 от каждого электрона ближайшей внутренней группы (s или p); 1,0 - от каждого электрона более глубоких слоёв (d или f). При этом нужно иметь в виду, что учитываются только электроны, находящиеся между ядром и исследуемой оболочкой. Электроны, внешние по отношению к исследуемому, в экранировании участия не принимают. При вычислении констант экранирования для данной электронной конфигурации необходимо вместо 1s, 2s, 2p и т.д. в правой части равенств подставить число электронов на данной орбитали. Радиальные функции слейтеровского типа недостаточно точно описывают поведение атомных орбиталей Хартри – Фока на небольших расстояниях от ядра. Этот недостаток можно устранить, если использовать для аппроксимации атомной орбитали Хартри – Фока по крайней мере две слейтеровские функции с разными орбитальными экспонентами:
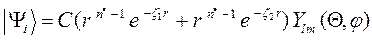
Двухэкспоненциальные функции дают хорошее приближение к функциям Хартри - Фока почти во всей области изменения  . Эти функции называют дубль – дзета функциями, а базисный набор, построенный на них – дубль – дзета - базисом (
. Эти функции называют дубль – дзета функциями, а базисный набор, построенный на них – дубль – дзета - базисом ( ). При расчёте волновых функций молекул широкое распространение получили функции гауссовского типа:
). При расчёте волновых функций молекул широкое распространение получили функции гауссовского типа:
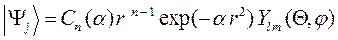
где 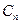 – нормировочный множитель;
– нормировочный множитель; 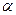 – варьируемый параметр. Эти орбитали более просты при квантово-химических расчётах, но они хуже аппроксимируют атомную орбиталь. Этот их недостаток компенсируется тем, что для описания одной атомной орбитали используют сумму нескольких гауссовых функций. Численные значения и
– варьируемый параметр. Эти орбитали более просты при квантово-химических расчётах, но они хуже аппроксимируют атомную орбиталь. Этот их недостаток компенсируется тем, что для описания одной атомной орбитали используют сумму нескольких гауссовых функций. Численные значения и 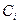 выбирают минимизацией энергии основного состояния атома с выбранным базисом. В заключение, рассмотрим, какие орбитали двух атомов могут комбинироваться между собой при образовании молекулы. Для того чтобы такая комбинация была эффективной, необходимо, чтобы:
выбирают минимизацией энергии основного состояния атома с выбранным базисом. В заключение, рассмотрим, какие орбитали двух атомов могут комбинироваться между собой при образовании молекулы. Для того чтобы такая комбинация была эффективной, необходимо, чтобы:
1. Энергии соответствующих атомных орбиталей были сравнимы по величине;
2. Их электронные облака, характеризующие плотность вероятности нахождения электрона вблизи ядра атома, по возможности полнее перекрывались;
3. Атомные орбитали обладали одинаковыми свойствами симметрии.
Если принять указанные приближения, то, используя только универсальные физические постоянные и не вводя никаких экспериментальных данных (разве только равновесные межъядерные расстояния, причём в последнее время всё чаще обходятся и без них), можно проводить чисто теоретические расчёты ab initio («от начала») по схеме метода самосогласованного поля. Такие расчёты ССП — ЛКАО — МО сейчас стали возможны уже для систем, содержащих несколько десятков электронов. Здесь основные трудности заключаются в том, что приходится вычислять громадное количество интегралов. Хотя подобные расчёты являются громоздкими и дорогостоящими, получающиеся результаты не всегда удовлетворительны, во всяком случае, с количественной стороны. Это объясняется тем, что, несмотря на различные усовершенствования схемы ССП — ЛКАО — МО (например, введение конфигурационного взаимодействия и др. способов учёта корреляции электронов), исследователи, в конечном счёте, ограничены возможностями одноэлектронного приближения ЛКАО — МО. В связи с этим, большое развитие получили полуэмпирические расчётные методы. Эти расчёты также восходят к уравнению Шрёдингера, но вместо того чтобы вычислять огромное количество (миллионы) интегралов, большую часть из них опускают (руководствуясь порядком их малости), а остальные упрощают. Потерю точности компенсируют соответствующей калибровкой параметров, которые берутся из эксперимента. Упрощение расчётов, в общем случае, идёт несколькими путями: понижение порядка детерминанта за счёт уменьшения числа атомных орбиталей, вовлекаемых в расчёт от каждого атома; упрощение детерминанта за счёт пренебрежения некоторыми интеграллами или замена их эмпирическими величинами. Такая методика, называемая полуэмпирической, позволяет при удачном выборе параметров компенсировать недостатки теоретической схемы, связанные с многочисленными пренебрежениями. Чаще всего в полуэмпирических методах используют валентное приближение, согласно которому при разложении молекулярных орбиталей в их линейную комбинацию – учитывают только валентные электроны и соответствующие им орбитали валентной оболочки. Внутренние электроны считают локализованными на соответствующих атомных орбиталях и образующими неполяризованный остов. Полуэмпирические расчёты пользуются большой популярностью, ибо оптимальным образом сочетают в себе простоту и точность в решении различных проблем. Описанные выше расчёты нельзя непосредственно сравнивать с чисто теоретическими (неэмпирическими) расчётами, так как у них разные возможности, а отсюда и разные задачи. Ввиду специфики используемых параметров при полуэмпирическом подходе нельзя надеяться получить волновую функцию, удовлетворительно описывающую различные (а тем более все) одноэлектронные свойства. В этом состоит коренное отличие полуэмпирических расчётов от расчётов неэмпирических, которые могут, хотя бы в принципе, привести к универсальной волновой функции. Поэтому сила и привлекательность полуэмпирических расчётов заключаются не в получении количественной информации как таковой, а в возможности интерпретации получаемых результатов в терминах физико-химических концепций. Только такая интерпретация и приводит к действительному пониманию, так как без неё на основании расчёта можно лишь констатировать те или иные количественные характеристики явлений (которые надёжнее определить на опыте). Именно в этой специфической особенности полуэмпирических расчётов и заключается их непреходящая ценность, позволяющая им выдерживать конкуренцию с полными неэмпирическими расчётами, которые по мере развития вычислительной техники становятся всё более легко осуществимыми. Что касается точности полуэмпирических квантово-химических расчётов, то она (как и при любом полуэмпирическом подходе) зависит скорее от умелой калибровки параметров, нежели от теоретической обоснованности расчётной схемы. Так, если выбирать параметры из оптических спектров каких-либо молекул, а затем рассчитывать оптические спектры родственных соединений, то нетрудно получить великолепное согласие с экспериментом, но такой подход не имеет общей ценности. Поэтому основная проблема в полуэмпирических расчётах заключается не в том, чтобы вообще определить параметры, а в том, чтобы одну группу параметров (например, полученных из оптических спектров) суметь использовать для расчётов других характеристик молекулы (например, термодинамических). Только тогда появляется уверенность, что работа ведётся с физически осмысленными величинами. При расчёте ненасыщенных соединений с кратными связями обычно используют так называемое π-электронное приближение, которое заключается в том, что вместо решения вариационной задачи для всех валентных электронов решают уравнения только для π-электронов, а σ-электроны считают неполяризованными и включают в ядерный остов молекулы. Этот метод позволяет достаточно просто, хотя и приближённо, рассчитывать сложные молекулы. Например, для этилена вместо 12 электронов квантово - механическая задача решается только для двух π-электронов. Для антрацена вместо 66 атомных орбиталей необходимо рассмотреть только 14. Число интегралов кулоновского отталкивания электронов можно резко сократить, используя приближение нулевого дифференциального перекрывания (НДП), введенное впервые Парром (1952). Приближение НДП используется в настоящее время во всех расчётных схемах и является почти универсальным. Такое приближение превращает четырехцентровые интегралы в двухцентровые, что значительно уменьшает время вычисления одного интеграла. Приближение НДП используется в расчётной схеме ППДП (полное пренебрежение дифференциальным перекрыванием), предложенной Поплом с сотрудниками (1965), и в ряде других вариантов этого полуэмпирического подхода, например ЧПДП – частичное пренебрежение дифференциальным перекрыванием, МЧПДП – модифицированное частичное пренебрежение дифференциальным перекрыванием и ОПДП – ограниченное пренебрежение дифференциальным перекрыванием. Метод ППДП и его разновидности являются в настоящее время наиболее разработанными полуэмпирическими методами, которые учитывают при расчёте молекул не только π-, но и σ-электроны. Наряду с относительно строгими и обоснованными полуэмпирическими методами (ППДП, ЧПДП, МЧПДП, ОПДП) получили развитие и использование, более простые методы расчёта. В этих методах пренебрегают всеми интегралами кулоновского отталкивания и компенсируют грубость этого приближения соответствующей параметризацией интегралов. Зачастую удаётся получать весьма убедительную информацию о строении и свойствах молекул без всяких громоздких расчётов, используя различные фундаментальные концепции, основанные главным образом на рассмотрении симметрии. Соображения симметрии играют важную роль, так как позволяют контролировать физический смысл результатов приближённого рассмотрения многоэлектронных систем. Например, исходя из точечной группы симметрии молекулы, можно вполне однозначно решить вопрос об орбитальном вырождении электронных уровней независимо от выбора расчётного приближения. Знание степени орбитального вырождения часто уже достаточно для суждения о многих важных свойствах молекулы, таких как потенциалы ионизации, магнетизм, конфигурационная устойчивость и ряд других свойств. Принцип сохранения орбитальной симметрии лежит в основе современного подхода к механизмам протекания согласованных химических реакций (правила Вудворда — Гофмана). Указанный принцип может быть, в конечном счёте, выведен из общего топологического рассмотрения областей связывания и антисвязывания в молекуле. В методе же валентных связей (МВС) полагают, что каждая молекула составлена из атомов, и для объяснения электронного строения молекул применяют атомные орбитали составляющих её атомов. Подобное приближение прямо противоположно методу молекулярных орбиталей, рассматривающему в своей наиболее общей форме каждую молекулу, как самостоятельное целое, а не простую совокупность атомов. Метод валентных связей рассчитан, прежде всего, на химиков, поскольку его наиболее проще приспособить к обычному для них образу мышления. В данном методе результаты работы Гайтлера и Лондона обобщены и распространены на многоатомные молекулы, для которых перенесены характерные особенности двухэлектронной связи в молекуле водорода. Принимается, что каждая связь осуществляется парой электронов с антипараллельными спинами. Такая электронная пара локализованной между двумя определёнными атомами, при этом атомные орбитали двух электронов перекрываются, причём последнее является не причиной, а следствием электростатического взаимодействия двух одноимённых зарядов. Представление о локализованной паре электронов является квантово-механическим аналогом более ранней идеи Льюиса о связи, как о паре электронов, в равной степени, принадлежащей двум атомам. Казалось бы, что метод валентных связей удобен для объяснения основных свойств ковалентной связи: её насыщаемости, направленности, поляризуемости, энергии активации химической реакции - однако в простом приближении данного метода, возникают большие трудности при описании электронного строения и свойств сопряжённых и ароматических соединений, а также неорганических молекул, причём не обязательно сложных. Если же к данному методу применить современные требования по расчёту молекулярных характеристик и истолкованию молекулярных спектров, трудности становятся весьма значительными. Сравнивая МВС м ММО, следует отметить, что достоинством первого является его наглядность: насыщаемость связи объясняется как максимальная ковалентность, направленность вытекает из направленности атомных и гибридных орбиталей; дипольный момент молекулы складывается из дипольных моментов связей, разности относительной электроотрицательности атомов, образующих молекулу, и наличия неподелённых электронных пар. Однако существование некоторых соединений невозможно объяснить с позиций МВС. Это электрон – дефицитные соединения и соединения благородных газов. Их строение легко объяснимо в рамках метода молекулярных орбиталей. Устойчивость молекулярных ионов и атомов в сравнении с молекулами легко предсказывается с позиций данного метода. И, наконец, магнетизм и окраска вещества также легко объясняются на основании метода молекулярных орбиталей. Количественные расчёты в методе молекулярных орбиталей, несмотря на свою громоздкость, все же гораздо проще, чем в методе валентных связей. В то же время качественно выводы метода валентных связей гораздо нагляднее и шире используются экспериментаторами, чем метода молекулярных орбиталей. В конечном счёте, выбор метода определяется объектом исследования и поставленной задачей. В высших своих приближениях и метод валентных связей, и метод молекулярных орбиталей приводят практически к одинаковым результатам, достигаемым, однако по-разному. Метод валентных связей более трудоёмкий и потому на сегодняшний день пользуется меньшим спросом, чем метод молекулярных орбиталей. В более же простом приближении, каждый из них обладает преимуществами в описании одних явлений и недостатками при описании других.

