 2015-04-20
2015-04-20 1454
1454Фотометричний метод заснований на визначенні ступеня светопоглинання розчинів залежно від концентрації забруднення за допомогою сучасних апаратів: абсорберів, фотоелектроколоріметров, спектрофотометрів.
Фотостабілізатори повинні володіти високою здатністю до світлопоглинання в діапазоні хвиль ультрафіолетового випромінювання між 300 і 400 нм (3000 і 4000 А) і відводити енергію в нешкідливої формі.
Ці методи складаються у вимірі поглинання променистої енергії розчинами аналізованих речовин. Характер спектра поглинання служить якісною ознакою обумовленої сполуки, а величина поглинання виступає як кількісна характеристика, що дозволяє судити про зміст компонента, що цікавить нас. Справа в тому, що поглинання променистої енергії за інших рівних умов пропорційно концентрації поглинаючої речовини.
Поняття фотометричні охоплює прийоми, які йменуються або йменувалися колориметричними, фотоелектроколориметричними, спектрофотометричними й властиво фотометричними. Походження цих термінів зв'язано зі способом виміру світлопоглинання й типом застосовуваного для цієї мети приладу. Однак в основі всіх способів лежить поглинання світла обумовленою речовиною у видимій, ультрафіолетовій і інфрачервоній області спектра.
По числу наукових публікацій фотометричний метод посідає перше місце. У дуже великому масштабі метод використовують у практиці аналізу найрізноманітніших об'єктів. Достоїнство цих прийомів у досить низькій межі виявлення, доступності й простоті, що сполучаються в багатьох випадках з достатньою вибірковістю й швидкістю; точність визначень також для ряду цілей цілком задовільна: відносна помилка звичайно становить
5 - 10%. Важливо й те, що фотометричні методи розроблені практично для всіх елементів і дуже багатьох органічних сполук.
Майже завжди виміру поглинання передує переклад обумовленого компонента в нову хімічну форму, що саме й відрізняє сильним поглинанням променистої енергії. Це може бути пофарбована сполука або сполуки, що поглинають випромінювання в ультрафіолетовій і інфрачервоній області спектра. Відшукання такої сполуки, вибір умов його утворення, знаходження прийомів усунення перешкод з боку інших компонентів - мета й істота хімічної теорії фотометричних методів. Найбільше поширення одержали прийоми, у яких використовують пофарбовані комплекси з органічними реагентами, наукові дослідження в основному присвячені саме цим комплексам, з'ясуванню хімізму відповідних реакцій.
У якості фотометричних реагентів використовують речовини різних класів. З неорганічних сполук - це галогеніди й роданіди, перекис водню, аміак, сполуки, що дають гетерополікислоти. З більш численних органічних реагентів можна назвати реагенти, що містять у певнім сполученні гідроксильну й карбоксильную групу, зокрема оксіазосполуки; реагенти з тіольної та тіонної групами. Дуже часто гарний аналітичний ефект дають багатокомпонентні сполуки, наприклад комплекси зі змішаною координаційною сферою.
Основними характеристиками електромагнітного випромінювання прийнято вважати частоту, довжину хвилі і поляризацію.
Довжина хвилі прямо пов'язана з частотою через (групову) швидкість поширення випромінювання. Групова швидкість поширення електромагнітного випромінювання у вакуумі дорівнює швидкості світла, в інших середовищах ця швидкість менше. Фазова швидкість електромагнітного випромінювання в вакуумі також дорівнює швидкості світла, в різних середовищах вона може бути як менше, так і більше швидкості світла.
Описом властивостей і параметрів електромагнітного випромінювання в цілому займається електродинаміка, хоча властивостями випромінювання окремих областей спектру займаються певні більш спеціалізовані розділи фізики (почасти так склалося історично, частково обумовлено істотною конкретною специфікою, особливо щодо взаємодії випромінювання різних діапазонів з речовиною, почасти також специфікою прикладних задач). До таких більш спеціалізованих розділів відносяться оптика (і її розділи) та радіофізика.
Жорстким електромагнітним випромінюванням короткохвильового кінця спектра займається фізика високих енергій, відповідно до сучасних уявлень, при високих енергіях електродинаміка перестає бути самостійною, об'єднуючись в одній теорії зі слабкими взаємодіями, а потім - при ще більш високих енергіях як очікується з усіма іншими калібрувальними полями.
Існують розрізняються в деталях і ступеня спільності теорії, що дозволяють змоделювати і дослідити властивості і прояви електромагнітного випромінювання. Найбільш фундаментальною із завершених і перевірених теорій такого роду є квантова електродинаміка, з якої шляхом тих чи інших спрощень можна в принципі отримати всі перераховані нижче теорії, що мають широке застосування в своїх областях. Для опису щодо низькочастотного електромагнітного випромінювання у макроскопічної області використовують, як правило, класичну електродинаміку, засновану на рівняннях Максвелла, причому існують спрощення в прикладних застосуваннях.
Для оптичного випромінювання (аж до рентгенівського діапазону) застосовують оптику (зокрема, хвильову оптику, коли розміри деяких частин оптичної системи близькі до довжин хвиль; квантову оптику, коли істотні процеси поглинання, випромінювання і розсіювання фотонів; геометричну оптику; граничний випадок хвильової оптики, коли довжиною хвилі випромінювання можна знехтувати). Гамма-випромінювання найчастіше є предметом ядерної фізики, з інших медичних і біологічних позицій вивчається вплив електромагнітного випромінювання в радіології. Існує також ряд областей фундаментальних і прикладних таких, як астрофізика, фотохімія, біологія фотосинтезу і зорового сприйняття, ряд областей спектрального аналізу, для яких електромагнітне випромінювання (найчастіше певного діапазону) і його взаємодія з речовиною відіграють ключову роль. Всі ці області межують і навіть перетинаються з описаними вище розділами фізики.
Деякі особливості електромагнітних хвиль c точки зору теорії коливань і понять електродинаміки:
· наявність трьох взаємно перпендикулярних (у вакуумі) векторів: хвильового вектора, вектора напруженості електричного поля E і вектора напруженості магнітного поля H.
· електромагнітні хвилі - це поперечні хвилі, в яких вектора напруженостей електричного і магнітного полів коливаються перпендикулярно до напрямку поширення хвилі, але вони істотно відрізняються від хвиль на воді і від звуку тим, що їх можна передати від джерела до приймача в тому числі і через вакуум.
Зако́н Бугера - Ламберта - Бера - физический закон, определяющий ослабление параллельного монохроматического пучка света при распространении его в поглощающей среде.
Закон выражается следующей формулой:
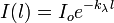 ,
,
где 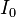 - интенсивность входящего пучка,
- интенсивность входящего пучка,
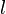 - толщина слоя вещества, через которое проходит свет,
- толщина слоя вещества, через которое проходит свет,
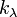 - показатель поглощения (не путать с безразмерным показателем поглощения
- показатель поглощения (не путать с безразмерным показателем поглощения 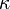 , который связан с
, который связан с 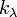 формулой
формулой 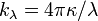 , где
, где 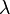 - длина волны).
- длина волны).
Показатель поглощения характеризует свойства вещества и зависит от длины волны λ поглощаемого света. Эта зависимость называется спектром поглощения вещества.
Фізичний закон, що визначає ослаблення паралельного монохроматичного пучка світла при поширенні його в поглинаючої середовищі.
Показник поглинання характеризує властивості речовини і залежить отдліни хвилі λ світла, що поглинається. Ця залежність називаетсяспектром поглинання речовини.
Відхилення від закону Бугера-Ламберта-Бера можуть бути також обумовлені недостатньою монохроматичністю пучка світла в приладі, флюоресценцією зразка або розсіюванням світла в розчині.
Порушення закону Бугера-Ламберта-Бера в результаті неправильної роботи приладу визначають по вимірюванню поглинання даного розчину в кюветах різної довжини.
Криві поглинання речовини при різних концентраціях ня відхилень від закону Бугера-Ламберта-Бера слід шукати в процесах, що відбуваються в розчині. При великих концентраціях спирту спостерігаються відхилення від закону Бугера-Ламберта-Бера, і проби необхідно розбавляти.
Концентрація спирту, при якій відсутня асоціація та виконується закон Бугера-Ламберта-Бера, зазвичай не перевищує 0,015 моль.
Закон Бугера - Ламберта - Бера экспериментально открыт французским учёным Пьером Бугером в 1729 году, подробно рассмотрен немецким учёным И.Г. Ламбертом в 1760 году и в отношении концентрации C проверен на опыте немецким учёным А. Бером в 1852 году.
Рефрактометрия (від латинського refraktus - переломлений і грецького metr - міряю, вимірюю) - це розділ прикладної оптики, у якому розглядаються методи виміру показника заломлення світла (n) під час переходу з однієї фази до іншої, чи, інакше кажучи, показник заломлення n - цей показник швидкостей світла що межують середовищах.
Рефрактометрия широко застосовується також і визначення будівлі координаційних сполук (комплексів молекулярного іхелатного типу), вивчення водневої зв'язку, ідентифікації хімічних сполук, кількісного і структурного аналізу, визначення физико - химических параметрів речовин.
У виробничої практиці показник заломлення світла n використовується контролю ступеня чистоти і порядку якості речовин; в аналітичних цілях - для ідентифікації хімічних сполук та його кількісного визначення. Отже, рефрактометрія - це метод дослідження речовин, заснований на визначенні показника заломлення (коефіцієнта рефракції) та деяких менших його функцій. З функцій n, які у хімії, найбільше значення мають: функція Лоренца -Ленца, похідна n за концентрацією розтворенних речовин (приріст n) і дисперсионні формули, які включають різниці показників заломлення обох довжин хвиль. Інкременти n використав рідинної хроматографії і за визначенні молекулярної маси полімерів методом розсіювання світла.
Рефрактометричний метод аналізу заснований на вимірюванні показника заломлення (n) речовини, що досліджується. При переході променя світла з одного оптично прозорого середовища в інше вія змінює свій первісний напрямок, тобто заломлюється.
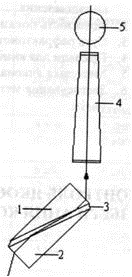 Рефрактометр (принципова схема)
Рефрактометр (принципова схема)
1 - вимірювальна призма,
2- освітлювальна призма,
3- рідина,
4- зорова труба,
5- окуляр
Відношення швидкості розповсюдження світла в вакуумі до швидкості розповсюдження світла в середовищі або відношення синусу кута падіння до синусу кута заломлення називається абсолютним показником заломлення; при переході променя світла з повітря у речовину - відносним показником заломлення іншого середовища по відношенню до першого.
Застосування рефрактометри в якісному аналізі:
Величину показника заломлення використовують в якісному аналізі для:
- ідентифікації речовин;
- визначення чистоти та тотожності речовин.
Застосування рефрактометрії в кількісному аналізі
Залежність показника заломлення від концентрації речовин в розчині покладена в основу кількісних визначень рефрактометричним методом і використовується для:
- визначення якості приготовлених розчинів та термінів зберігання
концентрованих розчинів;
- кількісного визначення компонентів в дво- та багатокомпонентних
сумішах.
Для рефрактометрического аналізу розчинів в широких діапазонах концентрацій користуються таблицями чи емпіричними формулами, найважливіші у тому числі (для розчинів сахарози, етилового спирту та інших.) затверджуються міжнародними угодами й у основі побудови шкал спеціалізованих рефрактометров для аналізу промислової власності й сільськогосподарської продукції. Розроблено способи аналізу трехкомпонентних розчинів, заснованих на виключно одночасному визначенні n і щільність чи в'язкості, або на здійсненні хімічних перетворень з виміром n вихідних і кінцевих розчинів; ці засоби застосовують при контролі нафтопродуктів, фармацевтичних препаратів та інших. Ідентифікація органічних сполук, мінералів, лікарських речовин здійснюється за таблицям n, процитованими в довідкових виданнях.
Перевагами рефрактометрического методу є її простота і щодо невисока вартість приладів визначення коефіцієнта заломлення світла.
Показник заломлення речовини - величина, що дорівнює відношенню фазових швидкостей світла (електромагнітних хвиль) в вакуумі і в даному середовищі 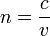 .
.
Також про показник заломлення говорять для будь-яких інших хвиль, наприклад, звукових.
Показник заломлення залежить від властивостей речовини і довжини хвилі випромінювання, для деяких речовин показник заломлення досить сильно змінюється при зміні частоти електромагнітних хвиль від низьких частот до оптичних і далі, а також може ще більш різко змінюватися в певних областях частотної шкали. За замовчуванням звичайно мається на увазі оптичний діапазон або діапазон, який визначається контекстом.
Існують оптично анізотропні речовини, в яких показник заломлення залежить від напрямку і поляризації світла. Такі речовини досить поширені, зокрема, це все кристали з досить низькою симетрією кристалічної решітки, а також речовини, піддані механічній деформації.
Показник заломлення можна виразити як корінь з добутку магнітної і діелектричної проникності середовища
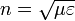
Треба при цьому враховувати, що значення магнітної проникності 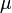 , та показника абсолютної діелектричної проникності
, та показника абсолютної діелектричної проникності  , для даного діапазону частот - наприклад, оптичного, можуть дуже сильно відрізнятися від статичного значення цих величин.
, для даного діапазону частот - наприклад, оптичного, можуть дуже сильно відрізнятися від статичного значення цих величин.
Для вимірювання коефіцієнта заломлення використовують ручні і автоматичні рефрактометри. При використанні рефрактометра для визначення концентрації цукру у водному розчині прилад називають Цукрометр.
Відношення синуса кута падіння (α) Променя до синуса кута заломлення (γ) При переході променя з середовища A в середу B називається відносним показником заломлення для цієї пари середовищ.
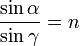
Величина n є відносний показник заломлення середовища В по відношенню до середовища А, а n '= 1 / n є відносний показник заломлення середовища А по відношенню до середовища В.
Ця величина, за інших рівних умов, зазвичай менше одиниці при переході променя з середовища більш щільною в середу менш щільну, і більше одиниці при переході променя з середовища менш щільною в середу більш щільну (наприклад, з газу або з вакууму в рідину або тверде тіло). Є винятки з цього правила, і тому прийнято називати середу оптично більш-менш щільною, ніж інша (не плутати з оптичною щільністю як мірою непрозорості середовища).
Луч, що падає з безповітряного простору на поверхню середовища В, заломлюється сильніше, ніж при падінні на неї з іншого середовища А; показник заломлення променя, що падає на середу з безповітряного простору, називається його абсолютним показником заломлення або просто показником заломлення даного середовища, це і є показник заломлення, визначення якого дано на початку статті. Показник заломлення будь-якого газу, в тому числі повітря, при звичайних умовах багато менше, ніж показники заломлення рідин або твердих тіл, тому наближено (і з порівняно непоганий точністю) про абсолютне показнику заломлення можна судити за показником заломлення щодо повітря.
Завдання кількісного аналізу - це визначення точного вмісту елементів йонів елементів йонів, або їх сполук досліджуваної речовини. Кількісний аналіз виконується хімічними і фізико-хімічними методами, їх відомо понад 50. Хімічні методи ґрунтуються на хімічних реакціях (охолодження і нейтралізації, окислення відновлення) до них відносять гравіметричні (вагові), титриметричні (об’ємні), оксиди метричні (окислення відновлення). Фізико-хімічні методи основані на вимірюванні фізичних речовин, хімічних реакцій в них можуть виконуватись, або не виконуватись і мати другорядне значення.
Кількісне визначення речовини хімічними методами складається з трьох етапів:
І - відмірювання певної кількості речовини для аналізу. Для цього речовину зважують чи вимірюють об’єм її розчину;
ІІ - проведення хімічної реакції, в результаті якої компонент, що визначають перетворюється у сполуку з певними хімічними і фізичними властивостями;
ІІІ - вимірювання показника якості фізичної властивості за величиною якого, роблять висновки про кількість компонента.
Важливе значення має правильний вибір умов проведення хімічної реакції.
І - Перша група методів кількісного хімічного аналізу включає методи, в основу яких покладено вимірювання показника властивості продукту реакції (Р). До першої групи методів належить гравіметричний і колориметричний аналізи;
ІІ - Друга група методів кількісного хімічного аналізу включає методи, в основі яких лежить вимірювання кількості реактиву витраченого на взаємодію з компонентом, що визначають. До цієї групи належать титриметричні (об’ємні) аналіз з його підрозділами. Основним є правильне визначення точки еквівалентності, тобто моменту коли між речовиною Х і реактивом R існує еквівалентне співвідношення.
За типом хімічної реакції розрізняють такі методи об'ємного аналізу: метод кислотно-основного титрування (нейтралізації), метод осадження і комплексоутворення, метод окислення відновлення (залежить від характеру реактиву, підрозділяється на перманганатометрію, йодометрію, хроматометрію).
ІІІ - До третьої групи методів кількісного хімічного аналізу відносять методи, які ґрунтуються на вимірюванні змін властивості самого компонента Х зумовлених зв’язуванням його реактивом R у певну хімічну сполуку. До них належать методи газового аналізу.
Основні операції гравіметричного аналізу: отримання осаду, відділення осаду, висушування, зважування. Вимоги до осадів:
- до основних операцій гравіметричного аналізу залежать:
- розрахунок наважки;
- взяття наважки;
- розчинення аналізуючої речовини;
- осадження;
- фільтрування;
- промивання осаду;
- висушування;
- прокалювання і зважування, розрахунок результатів.
Відбір проби: проба - це невелика кількість речовини взята для аналізу. Для аналізу беруть середню пробу, яка складається із багатьох проб взятих у різних точках. Правильно відібрана проба дає правильні результати.
Зважування: для приблизного зважування з точністю до 1-0,1 г використовують технохімічні ваги, а для точного зважування використовують аналітичні ваги з точністю до четвертого знака після коми до 0,0001 г. Марки аналітичних терезів: ВЛАО 200, ВАДП 100. Максимальна маса, яку можна зважити 100-200 г.
Взяття наважки: наважка - це невелика кількість речовини взята для аналізу. Речовину не можна зважувати на чашках терезів. Для зважування речовини поміщають в бюкс чи на часове скло. Відомо два способи взяття наважки: І - наважку поміщають в попередньо зважену тару, зважують і по різниці мас обчислюють масу наважки, яку необхідно повністю перенести наважку в склянку. ІІ - посудину для зважування насипають аналізуючи речовину, зважують разом із посудиною, висипають у склянку зважують для розчинення, а посудину із залишками знову зважують по різниці мас визначають величину наважки.
Наважка, яка береться для аналізу неповинна бути дуже малою тому, що збільшується помилка аналізу, неповинна бути дуже великою тому, що фільтрування, сушка, прожарювання займуть багато часу. Встановлено, що для утворення кристалічних осадів наважку треба взяти біля 0,5 г, а для аморфних 0,1-0,3 г.
Відбір осаджувача і його концентрація. На практиці осаджувача беруть в 1,5-2 рази більше ніж потрібно за рівнянням надлишок осаджувача зменшує розчинність осаду. Для осадження кристалічних осадів користуються розведеними розчинниками. В цих умовах осади утворюються у формі більших кристалів. Аморфні осади осаджують із концентрованих розчинів.
Умови отримання крупно кристалічних і аморфних осадів
Для отримання крупнокристалічних осадів необхідно дотримуватись таких умов садження:
- осаджують із достатньо розбавленого розчину розбавленим розчином.
- розчин осаджувача добавляють повільно, краплями, особливо на початку осадження.
- при введені осаджувача розчин потрібно добре перемішувати скляною паличкою;
- осадження проводять із гарячого розчину гарячим розчином осаджувача, що сприяє росту кристалів.
- осад залишають на декілька годин на дозрівання, що веде до укруплення кристалів.
Осадження аморфних осадів проводиться за таких умов:
- осадження із гарячих розчинів;
- перед осадженням у розчин добавляють коагулюючий електроліт.
- осадження проводять із концентрованих розчинів концентрованими розчинами осаджувача, осаджувач вводять швидко;
- аморфні осади фільтрують і промивають відразу ж після осадження без дозрівання.
Після осадження проводять пробу на повноту осадження: в розчин з відстояним осадом обережно по стінці стакана приливають декілька крапель осаджувача і слідкують за місцем стікання крапель, якщо в місті падіння крапель осаджувача в розчині не утворилось муті це означає, що осадження відбулося повністю. Фільтрування і промивання осаду важливі операції вагового аналізу. Фільтрують через паперові беззольні фільтри у яких зола, що залишилась після спалювання настільки незначна, що нею можна знехтувати. Беззольні фільтри розрізняють за діаметром (5, 7, 9, 11 см) і щільністю паперу (від щільності фільтра залежить швидкість фільтрування). Вони бувають трьох сортів: І - найменшої щільності, що швидко фільтрують, їх застосовують при фільтруванні аморфних осадів (чорна, або червона стрічка); ІІ - середньої щільності, використовуються для фільтрування кристалічних осадів (біла стрічка); ІІІ - найбільш щільні, які фільтрують повільно і використовуються при фільтруванні мілко-кристалічних осадів (синя стрічка).
При фільтруванні спочатку зливають відстояну рідину над осадом через фільтр по щільно приставленій до носика стакана скляній паличці. Рівень рідини на фільтрі повинен бути нижче краю фільтра на 3-5 мм. Коли вся рідина злита з осаду і на дні стакана залишився осад приступають до промивання осаду шляхом декантації. Кількість промивної рідини залежить від характеру осаду, аморфні осади промиваються довше ніж кристалічні, при цьому промивну рідину потрібно вливати малими порціями. Промивають осад декантацією до тих пір поки стікаюча рідина не буде мати домішок, для цього збирають в пробірку 1-2 мл фільтрату, і приливають реактив, який дає якісну реакцію на домішки (хлориди - з AgNO3, утворюється біла муть, якщо нема муті то в осаді немає домішок). Далі осад разом із фільтром висушують при температурі 100-110оС і прожарюють в муфельній печі при високій температурі.
До методів кількісного аналізу належать:
1) гравіметричний (ваговий) метод, який ґрунтується на точному вимірюванні маси речовини, що визначається, або її складових частин, що виділяються в чистому стані або у вигляді відповідних сполук точно відомого постійного складу;
2) титриметричний (об’ємний) метод, в якому кількість досліджуваної речовини визначається шляхом точного вимірювання об’ємів реагуючих речовин.
Гравіметричний (ваговий) аналіз - полягає в тому, що складову частину речовини визначають відокремлюючи осадження, у вигляді осаду, відомого сталого складу, а потім зважують. Осад, що утворився повинен відповідати таким вимогам:
- бути практично нерозчинним;
- склад осаду після висушування, чи прожарювання повинен відповідати певній формі;
- розмір зерен повинен бути більший ніж пори фільтра (осад повинен добре відокремлюватись від фільтрату);
- молярна маса вагової форми повинна бути по можливості великою.
Розрізняють три варіанти вагового аналізу: визначення вологи, визначення золи, визначення за масою осаду.
Визначення вологи: точну наважку речовини висушують, волога випаровується маса зменшування і по зменшенню маси визначають вміст вологи:
W = m1 – m2/ m1 * 100%
Визначення золи: спалюють в тиглі точну наважку аналізуючої речовини, прокалюють залишок до тих пір поки маса перестане зменшуватись тобто до постійної маси. Залишок золи зважують і обчислюють вміст золи в аналізюючій речовині:
W = m1/m * 100%
m1 – маса золи,
m – маса наважки
Визначення по масі осаду:аналізуючи речовину за допомогою хімічної реакції переводять в осад, який фільтрують, промивають, висушують, зважують і по масі його визначають вміст аналізуючої речовини. Цей спосіб найбільш поширений.
Переваги і недоліки гравіметричного аналізу: Не потрібно складного обладнання, але метод є не дуже точним і виконується довго, не дивлячись на це має найбільш широке використання.
Титрометричний аналіз (титрування) - методи кількісного аналізу, основані на зміні об’єму розчину реактиву точно відомої концентрації, що витрачається для реакції с визначеною речовиною.
Титрування - процес визначення титру дослідницької речовини.
Титрування виготовлюють за допомогою бюретки, заповненої титр антом до нульової відмітки. Титрування починають з інших відміток, не рекомендовано, так як школа бюретки може бути нерівномірною.
Заповнення бюреток робочим розчином виробляють через воронку або з допомогою спеціальних пристосувань, якщо бюретка полу автоматична. Кінцеву точку титрування (точку еквівалентності) визначають індикаторам або фізико-хімічним (по електропровідності, потенціалу індикаторного електроду тощо). По кількості робочого рас твору, що пішло на титрування розраховують результати аналізу
Нефелометричний і турбідиметричний методи аналізу базуються на тому, що компонент, який визначається, переводять у малорозчинну сполуку, що знаходиться у вигляді суспензії і здатна розсіювати світло.
При проходженні пучка світла через суспензію дрібних частинок у розчиннику (дисперсійна система) відбувається бічне розсіювання світла (мутність, що візуально спостерігається).
Якщо довжина хвилі менша лінійних розмірів частинок, то розсіювання обумовлене переломленням світла на границі розділу частинка - розчинник і відбиттям його частинками. Якщо довжина хвилі більша лінійних розмірів частинок, то відбувається дифракція світлової хвилі, виникає ефект Тіндаля. Інтенсивність розсіювання зростає зі збільшенням числа частинок, що розсіюють. При нефелометричних визначеннях вимірюють інтенсивність розсіювання світла Wp у напрямку, перпендикулярному до напрямку первинного пучка світла.
Нефелометричною суспензією називають суспензії малорозчинних речовин при їхньому вмісті 100 мг/дм3 і менше.
Метод визначення вмісту речовин за інтенсивністю (потужністю) розсіювання світла W0 називається нефелометрією.
Метод визначення вмісту речовин за інтенсивністю (потужністю) ослаблення основного світлового потоку W називається турбідиметрією.
Частинки відбивають постійну частину світла за інтервал часу, достатній для виміру. Потужність світлового потоку, що розсіюється дрібними частинками суспензії, описується рівнянням Релея:
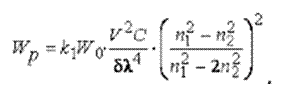
де k1 - коефіцієнт; V - середній об’єм частинки суспензії; C - концентрація частинок; 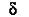 - густина речовини частинки;
- густина речовини частинки; 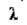 - довжина хвилі падаючого пучка світла; n1 - показник заломлення частинок суспензії; n1 - показник переломлення розчинника.
- довжина хвилі падаючого пучка світла; n1 - показник заломлення частинок суспензії; n1 - показник переломлення розчинника.
Величини 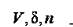 - залежать від умов одержання і вимірювання: швидкості змішування реагентів, швидкості перемішування, температури, кислотності середовища, присутності сторонніх іонів, довжини хвилі падаючого світла. При сталості умов величини
- залежать від умов одержання і вимірювання: швидкості змішування реагентів, швидкості перемішування, температури, кислотності середовища, присутності сторонніх іонів, довжини хвилі падаючого світла. При сталості умов величини  є постійними. Поєднуючи усі постійні величини в одну константу, одержимо:
є постійними. Поєднуючи усі постійні величини в одну константу, одержимо:
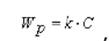
тобто потужність розсіяного світлового потоку прямо пропорційна концентрації частинок суспензії.
Для двох каламутних середовищ з частинками однакової форми і розмірів відношення інтенсивності розсіяного світла пропорційно концентрації частинок:
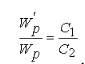 , звідси
, звідси 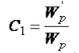 (3.22)
(3.22)
Рівняння (3.22) використовується при нефелометричних визначеннях. Нефелометричні визначення мають чутливість, порівняну з фотометричними визначеннями.
Нефелометр - прилад, що має пристрій для спостереження розсіяного світла під кутом 90˚С до напрямку падаючого променя. Він призначений для визначення концентрацій колоїдних розчинів, суспензій і емульсій. Принцип дії приладу заснований на вирівнюванні світлових потоків: одного, що розсіюється частинками суспензії, іншого - від матового або молочного скла розсіювача. Потоки зрівнюють зміною потужності одного з потоків за допомогою діафрагми. Для визначення концентрації суспензії будують градуювальний графік.
У нефелометрії і турбідиметрії використовують ті ж методи, що і у фотометрії, а саме: метод калібрувального графіка, метод порівняння, метод добавок.
При нефелометричних і турбідиметричних аналізах необхідно дотримуватись умов, які визначають успішність роботи:
1. Внаслідок того, що при роботі цим методом звичайно використовують сильно розведені розчини, осади, що одержуються, повинні мати дуже маленьку розчинність;
2. Як видно з вище наведених рівнянь, значення інтенсивності розсіяного і поглиненого світла залежать від розмірів частинок, що містяться в розчині. Тому одержання правильних результатів при аналізі суспензій залежить від методики одержання суспензій і від відтворюваності їх оптичних властивостей.
На розміри частинок і оптичні властивості суспензії впливають наступні фактори:
- концентрація іонів, які утворюють осад;
- відношення між концентраціями розчинів, які змішуються;
- порядок змішування розчинів;
- швидкість змішування;
- час, який потрібен для одержання максимальної мутності;
- стабільність дисперсії;
- присутність сторонніх електролітів;
- присутність неелектролітів;
- температура;
- наявність захисних колоїдів.
Таким чином, вивчення всіх цих факторів і стандартизація умов підготовки речовини до нефелометричного визначення необхідні для правильної роботи.
3. Суспензії повинні бути стійкими у часі, тобто не осідати протягом достатньо тривалого часу. Для збільшення стійкості суспензій часто використовують захисні колоїди. Всі ці обмеження призводять до того, що нефелометричні і турбідиметричні методи виявляються менш точними, ніж фотометричні. В практиці аналітичної хімії вони використовуються тільки в тих випадках, коли визначувані іони або речовини неможливо визначити фотометричними методами, наприклад, сульфати і хлориди, які не дають стійких забарвлених сполук. Для точних нефелометричних досліджень використовують нефелометри, які побудовані по принципу візуальних або фотоелектричних колориметрів. В деяких випадках турбідиметричні визначення проводять методом стандартних серій. Але необхідність створення постійних умов визначення роблять цей метод досить неточним, напівкількісним.
Достатньо широко використовується метод турбідиметричного титрування. При цьому можна використовувати тільки такі реакції, які протікають швидко, наприклад реакції утворення аргентум хлориду або барій сульфату, і не можна використовувати реакції, проведення яких потребує складних операцій.
Трубідиметрія (лат. turbidus - мутний) - метод кількісного аналізу, що базується на вимірюванні інтенсивності світла, яке поглинається суспензією твердої речовини (джерело і приймач випромінювання знаходяться на одній лінії). При турбідиметричних вимірюваннях величину, яку називають мутністю (S), визначають за рівнянням, яке має вигляд, аналогічний рівнянню Бугера - Ламберта - Бера:
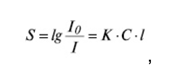
де I та I0 - інтенсивність світлового потоку, який пройшов крізь суспензію, що падає на неї, відповідно; K - молярний коефіцієнт мутності розчину; l - товщина шру розчину, що поглинає; C - концентрація часток, що поглинають у розчині.
Для турбідиметричних вимірювань придатні колориметри, фотоколориметри, спектрофотометри (як правило, використовують синій світлофільтр). Порядок вимірювань збігається з порядком колориметричних вимірювань. Кількісні визначення проводять з використанням калібрувальної кривої. Турбідиметричний метод має високу чутливість, але його застосування обмежене у зв’язку з тим, що на світлорозсіювання дуже впливає розмір часток, тому при порівнянні стандартів і проби необхідне суворе дотримання ідентичності умов. Труднощі, пов’язані із впливом різних факторів на властивості суспензій, зменшуються при фототурбідиметричному титруванні, коли точку еквівалентності визначають за максимумом помутніння.
Фототурбідиметричне титрування можна виконувати візуально або з використанням абсорбційного фотометра.
Поляриметричний метод розроблено й використовується для якісного визначення вищенаведених речовин, при здійсненні контролю якості цукру-піску, цукру-рафінаду, кондитерських виробів, молочних продуктів, вина.
Визначення проводять з допомогою приладу поляриметра чи цукрометра.
З допомогою цього методу також вивчається структура речовин й наявність патологічних мікроорганізмів чи біологічно активних продуктів.

