2014-02-12
2014-02-12 1036
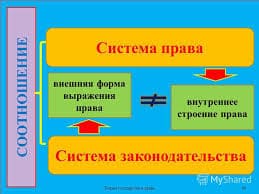
1036Рис. 3. Профиль поверхности потенциальной энергии вдоль координаты реакции.
Вертикальные штриховые линии ограничивают область активированного комплекса на координате реакции размером δ, горизонтальные сплошные линии – нулевые колебательные энергии реагентов и комплекса.
Доля активированных комплексов, переходящих через вершину барьера в единицу времени, равна 1/τ. Следовательно, если с# представляет собой число активированных комплексов в единице объема, находящихся в пределах отрезка δ, то величина с#/τ равна числу комплексов переходящих через вершину барьера за единицу времени в единице объема. Индекс «#» принято относить к активированному комплексу. Если каждый комплекс, переходящий через барьер, распадается, то величина c#/τ равняется скорости реакции r, т. е.
r = c# / τ = c# (kT/2πm*)1/2 ∙ 1/δ = kcA∙cB…., (6)
где сA, cB,…. – концентрации молекул реагентов, следовательно, константа скорости реакции
k = c# / cA ∙ cB…∙(kT/2πm*)1/2 ∙ 1/δ. (7)
Согласно сделанному предположению, активированный комплес находится в равновесии с исходными веществами. Для такой системы можно написать константу равновесия в виде:
K# = c# / cA ∙ cB… (8)
Кроме того, согласно статистической термодинамике, константу равновесия реакции А + В + … можно выразить через статистические суммы состояний (или функции распределения)
K# = F# / FA ∙ FB… (9)
где F – полные статистические суммы состояний на единицу объема.
Статистическая сумма является термодинамической величиной, представляющей собой вероятность того, что молекула находится в данном поступательном, вращательном, колебательном, электронном состоянии.
Полная статистическая сумма на единицу объем
F = Fпост. Fвращ.Fколеб.Fэлектр. (10)
При обычных для каталитических реакций температурах колебательные суммы не сильно отличаются от единицы и для расчета F нужно учитывать Fпост. и Fвращ.
С помощью статистической суммы можно рассчитывать все термодинамические величины: константы равновесия, свободные энергии, энтропии и др. При расчете статистической суммы можно вынести из Fколеб. уравнения (10) множитель, содержащий разность энергий нулевых колебаний (уровней при абсолютном нуле температур, см. рис. 3), и представить константу равновесия в виде:
K# = F#1 / FA ∙ FB…∙ e-Eo/RT, (11)
где Eo – разность между энергиями нулевых уровней, приходящихся на 1 моль активированного комплекса и исходных веществ. Это величина энергии при абсолютном нуле, которую должны получить реагенты, чтобы перейти в активированное состояние и прореагировать; т. е. это энергия активации реакции при абсолютном нуле температур.
Из уравнения (7), (8) и (11) следует, что
k = F#1 / FA ∙ FB…∙(kT/2πm*)1/2 ∙ 1/δ ∙ e-Eo/RT, (12)
Вместо величины F#1 , представляющей полную статистическую сумму активированного комплекса, удобно пользоваться новой суммой – F#, в которую не входит множитель F#пост(1), соответствующий степени свободы поступательного движения по координате реакции, такой, что
F#1 = F# F#пост(1).
Сумма состояний поступательного движения, согласно статистической термодинамике:
F#пост(1) = (2πm*∙ kT/)1/2 / h ∙ δ (13)
Отсюда получаем следующее выражение для константы скорости:
k = kT/h ∙ F#1 / FA ∙ FB…∙ e-Eo/RT (14)
В уравнении (14) величина kT/ h является одинаковой для всех веществ и для всех реакций, поэтому множитель kT/h, имеющий размерность частоты, представляет для каждой температуры универсальную постоянную – частоту, с которой активированый комплекс переходит через вершину барьера. При 25 0С эта частота равна 6∙1012 с-1.
В окончательную расчетную формулу вводят еще трансмиссионный коэффициент χ:
k = χ ∙ kT/h ∙ F#1 / FA ∙ FB…∙ e-Eo/RT (15)
Трансмиссионный коэффициент χ учитывает возможность того, что не каждый активированный комплекс, достигший вершины барьера и движущийся по координате реакции, действительно будет распадаться и давать продукты реакции.
Он близок к единице для адиабатических процессов, т. е. таких процессов, в которых перестройка электронной оболочки при изменении координат ядер успевает осуществиться.
Если электронные реорганизации не успевают произойти, такие процессы называют неадиабатическими и для них χ < 1, причем могут быть значения χ, равные 10-5 – 10-8.
Неадиабатические процессы происходят главным образом при изменении ориентации спинов электронов, если последнее необходимо для осуществления процесса.
Уравнения (9), (15) позволяют выразить константу скорости через величину
k = χ ∙(kT/h) ∙ K# = χ ∙ (kT/h) ∙ e-ΔGo/RT (16)
где ΔGo – стандартное значение свободной энергии Гиббса при образовании активированного комплекса. Учитывая зависимость ΔG от изменения энтропии ΔS и энтальпии ΔH
ΔG = ΔH - T ΔS, (17)
получаем
k = χ ∙ (kT/h) ∙ exp (ΔS#/R) exp (-ΔH#/RT). (18)
Величина ΔH# равнозначна энергии активации, энтропия активации ΔS# может быть положительной или отрицательной. Как видно из уравнения (18), чем больше энтропия активации, тем выше скорость реакции, поэтому выгодна наименьшая упорядоченность активированного комплекса.
Уравнение (15) является основой для расчета предэкспоненциальных множителей в уравнении Аррениуса. Трудности при расчете связаны главным образом с выбором наиболее вероятной конфигурации активированного комплекса. Чаще всего принимают его конфигурацию близкой по строению к исходным реагентам или конечным продуктам.
Некоторым общим указанием по выбору конфигураций активированных комплексов является сформулированный Ф Райсом, и Э. Теллером принцип наименьшего действия – в реагирующей системе образование конечных продуктов должно протекать с наименьшими возможными перемещениями атомов и электронов.
Метод активированного комплекса в области адсорбции и катализа был впервые применен М.И. Темкиным в 1938 г. Активированный комплекс принимается адсорбированным на поверхности с одной существенной особенностью, она заключается в том, что необходимо учитывать возможность занятия нескольких центров поверхности одним активированным комплексом. Кроме того, в отличие от гомогенных реакций, активированные комплексы на поверхности, как правили, не обладают поступательным и вращательным движением. Концентрация активированных комплексов на поверхности должна зависеть от числа центров, занятых активированным комплексом, от числа способов осуществления конфигурации активированного комплекса, от его структуры, структуры поверхности, параметра решетки твердого тела, размеров молекулы, общего числа центров на поверхности.
Число активных центров обычно значительно меньше, чем число атомов поверхности (1015 см-2), при этом на металлах число активных центров сравнительно велико и приближается к числу поверхностных атомов металла, например, для гидрирования бензола на Pt, Pd, Ni это число составляет 1014 см-2.