2015-10-13
2015-10-13 6326
6326Зачет по медицинской химии. Вопросы билетов по компьютерному молекулярному моделированию и методам QSAR.
Общие сведения
Аббревиатура QSAR является сокращением от английского Quantitative Structure Activity Relationships, что в переводе на русский язык обозначает Количественное Cоотношение Cтруктура Активность (поэтому иногда в русскоязычной литературе используют сокращение КССА).
Одной из важнейших задач современной химической науки является установление зависимостей между структурой и свойствами веществ. Число вновь синтезируемых новых органических соединений постоянно увеличивается поэтому самой актуальной задачей является количественное предсказание конкретных свойств для новых еще не синтезированных веществ на основании определенных физико-химических параметров отдельных соединений.
Исторически всё началось с попыток учёных найти количественную связь между структурами веществ и их свойствами и выразить эту связь в количественном виде, например в виде математического уравнения. Это уравнение должно отражать зависимость одного числового набора (представляющих свойства) от другого числового набора (представляющего структуры). Выразить в числовом виде свойство достаточно просто – физиологическую активность серии веществ можно измерить количественно. Гораздо сложнее численно выразить структуры химических соединений. Для такого выражения в настоящее время в QSAR используются так называемые дескрипторы химической структуры.
Дескриптор – параметр, характеризующий структуру органического соединения, причём так, что подмечаются какие-то определенные особенности этой структуры. В принципе дескриптором может являться любое число, которое можно рассчитать из структурной формулы химического соединения – молекулярный вес, число атомов определенного типа (гибридизации), связей или групп, молекулярный объём, частичные заряды на атомах и т.д.
Для предсказания физиологической активности в QSAR обычно используют дескрипторы, рассчитанные на основе стерических, топологических особенностей структуры, электронных эффектов, липофильности. Значительную роль в QSAR имеют так называемые топологические дескрипторы. Структурные дескрипторы играют важную роль при оценке прочности связывания исследуемого соединения с молекулой-биомишенью, дескрипторы электронных эффектов описывают ионизацию или полярность соединений. Дескрипторы липофильности позволяют произвести оценку способности растворяться в жирах, то есть характеризует способность лекарства преодолевать клеточные мембраны и разного рода биологические барьеры.
В методе QSAR структурная формула представляется в виде математического представления - графа и оперируется с помощью специализированного математического аппарата - теории графов. Граф - математический объект, заданный множеством вершин и набором упорядоченных или неупорядоченных пар вершин (ребер). Теория графов позволяет посчитать так называемые инварианты графов, которые и рассматриваются как дескрипторы. Применяются также и сложные фрагментные дескрипторы, которые оценивают вклад различных частей молекулы в общее свойство. Они значительно облегчают исследователям обратное структурное конструирование неизвестных соединений с потенциально высокой активностью. Таким образом модель QSAR - это математическое уравнение (модель), с помощью которого можно описать как физиологическую активность (частный случай), так и вообще любое свойство, и этом случае правильнее говорить о QSPR - количественном соотношении между структурой и свойством.
Методология QSAR работает следующим образом. Сначала группу соединений с известной структурой и известными значениями физиологической активности (полученными из эксперимента) делят на две части: тренировочный и тестовый набор. В этих наборах цифры, характеризующие активность, уже соотнесены с конкретной структурой. Далее выбираются дескрипторы (в настоящее время придуманы многие сотни дескрипторов, однако реально полезных достаточно ограниченное число; существуют разные подходы к выбору наиболее оптимальных дескрипторов). На следующем этапе строят математическую зависимость (подбирают математическое уравнение) активности от выбранных дескрипторов для соединений из тренировочного (обучающего) набора и в итоге получают так называемое QSAR-уравнение,
Правильность построенного уравнения QSAR проверяют на тестовом наборе структур. Сначала вычисляют дескрипторы для каждой структуры из набора тестовой выборки, затем подставляют их в QSAR-уравнение, рассчитывают значения активности и сравнивают их с уже известными экспериментальными значениями. Если для тестового набора наблюдается хорошее совпадение расчётных и экспериментальных значений, то данное QSAR-уравнение можно применить для предсказания свойств новых, ещё не синтезированных структур. Метод QSAR позволяет, имея в распоряжении совсем небольшое количество химических соединений с известной активностью, предсказать необходимую структуру (или указать направления для модификация) и тем самым резко ограничить круг поисков.
В развитых странах работы в области QSAR ведутся постоянно возрастающими темпами - применение методов QSAR при создании новых соединений с заданными свойствами позволяет значительно сократить время и ресурсы и осуществлять более целенаправленный синтез соединений, обладающие необходимым заданных комплексом свойств.
Вопрос №3. Понятие о молекулярных графах и их инвариантах. Типы дескриптором молекулярной структуры. Понятие о топологических индексах. Индексы Винера, Рандича, Кира-Холла и другие топологические индексы. QSAR с использованием топологических индексов.
Молекулярный граф — связный неориентированный граф, находящийся во взаимно-однозначном соответствии со структурной формулой химического соединения таким образом, что вершинам графа соответствуют атомы молекулы, а рёбрам графа — химические связи между этими атомами. Понятие «молекулярный граф» является базовым для компьютерной химии и хемоинформатики. Как и структурная формула, молекулярный граф является моделью молекулы, и как всякая модель, он отражает далеко не все свойства прототипа. В отличие от структурной формулы, где всегда указывается, к какому химическому элементу относится данный атом, вершины молекулярного графа могут быть непомеченными — в этом случае молекулярный граф будет отражать только структуру, но не состав молекулы. Точно так же рёбра молекулярного графа могут быть непомеченными — в таком случае не будет делаться различие между ординарными и кратными химическими связями. В некоторых случаях может использоваться молекулярный граф, отражающий только углеродный скелет молекулы органического соединения. Такой уровень абстрагирования удобен для вычислительного решения широкого круга химических задач.
Естественным расширением молекулярного графа является реакционный граф, рёбра которого соответствуют образованию, разрыву и изменению порядка связей между атомами.
«Подчеркнём, что именно в теории Р. Бейдера впервые нашла обоснование эмпирическая идея аддитивности, именно эта теория позволила придать строгий физический смысл целому ряду понятий классической теории химического строения, в частности, „валентному штриху“ (связевый путь) и структурной химической формуле (молекулярный граф).»
Топологический индекс — инвариант (инвариант - термин, обозначающий нечто неизменяемое) молекулярного графа в задачах компьютерной химии. ЭЭто некоторое (обычно числовое) значение (или набор значений), характеризующее структуру молекулы. Обычно топологические индексы не отражают кратность химических связей и типы атомов (C,N,O и.т.д.), атомы водорода не учитываются. К наиболее известным топологическим индексам относятся индекс Хосои, индекс Винера, индекс Рандича, индекс Балабана и другие.
Глобальные и локальные индексы
Индекс Хосои и индекс Винера — примеры глобальных (или интегральных) топологических индексов, отражающих структуру данной молекулы. Бончев и Полянский предложили локальный (дифференциальный) индекс для каждого атома в молекуле. В качестве другого примера локальных индексов можно привести модификации индекса Хосои.
Дискриминирующая способность и супериндексы
Значения одного и того же топологического индекса для нескольких разных молекулярных графов могут совпадать. Чем меньше таких совпадений — тем выше так называемая дискриминирующая способность индекса. Эта способность является важнейшей характеристикой индекса. Для ее повышения несколько топологических индексов могут быть объединены в один супериндекс.
Вычислительная сложность является другой важной характеристикой топологического индекса. Многие индексы, такие как индекс Винера, индекс Рандича и индекс Балабана вычисляются с помощью быстрых алгоритмов, в отличие, например, от индекса Хосои и его модификаций, для которых известны только экспоненциальные по времени алгоритмы.
Применение
Топологические индексы используются в компьютерной химии для решения широкого круга общих и специальных задач. К этим задачам относятся: поиск веществ с заранее заданными свойствами (поиск зависимостей типа «структура-свойство», «структура-фармакологическая активность»), первичная фильтрация структурной информации для бесповторной генерации молекулярных графов заданного типа, предварительное сравнение молекулярных графов при их тестировании на изоморфизм и ряд других. Топологический индекс зависит только от структуры молекулы, но не от ее состава, поэтому молекулы одинаковой структуры (на уровне структурных формул), но разного состава, например, фуран и тиофен будут иметь равные индексы. Для преодоления этого затруднения был предложен ряд индексов, например, индексы электроотрицательности.
При векторном описании химической структуре ставится в соответствие вектор молекулярных дескрипторов, каждый из которых представляет собой инвариант молекулярного графа.
Молекулярные дескрипторы. Типы молекулярных дескрипторов.
Существующие наборы молекулярных дескрипторов могут быть условно разделены на следующие категории:
1. Фрагментные дескрипторы существуют в двух основных вариантах — бинарном и целочисленном. Бинарные фрагментные дескрипторы показывают, содержится ли данный фрагмент (подструктура) в структурной формуле, то есть содержится ли данный подграф в молекулярном графе, описывающем данное химическое соединение, тогда как целочисленные фрагментные дескрипторы показывают, сколько раз данный фрагмент (подструктура) содержится в структурной формуле. То есть сколько раз содержится данный подграф в молекулярном графе, описывающем данное химическое соединение. Уникальная роль фрагментных дескрипторов заключается в том, что, они образуют базис дескрипторного пространства, то есть любой молекулярный дескриптор (и любое молекулярное свойство), являющийся инвариантом молекулярного графа, может быть однозначно разложен по этому базису. Кроме моделирования свойств органических соединений, бинарные фрагментные дескрипторы в форме молекулярных ключей (скринов) и молекулярных отпечатков пальцев применяются при работе с базами данных для ускорения подструктурного поиска и организации поиска по подобию.
2. Топологические индексы. (информацию по ним см. выше)
3. Физико-химические дескрипторы — это числовые характеристики, получаемые в результате моделирования физико-химических свойств химеческих соединений, либо величины, имеющие четкую физико-химическую интерпретацию. Наиболее часто используются в качестве дескрипторов: липофильность (LogP), молярная рефракция (MR), молекулярный вес (MW), дескрипторы водородной связи[7], молекулярные объемы и площади поверхностей.
4. Квантово-химические дескрипторы — это числовые величины, получаемые в результате квантово-химических расчетов. Наиболее часто в качестве дескрипторов используются: энергии граничных молекулярных орбиталей (ВЗМО и НСМО), частичные заряды на атомах и частичные порядки связей, индексы реакционной способности Фукуи (индекс свободной валентности, нуклеофильная и электрофильная суперделокализуемость), энергии катионной, анионной и радикальной локализации, дипольный и высшие мультипольные моменты распределения электростатического потенциала.
5. Дескрипторы молекулярных полей — это числовые величины, аппроксимирующие значения молекулярных полей путем вычисления энергии взаимодействия пробного атома, помещенного в узел решетки, с текущей молекулой. На построении корреляций между значениями дескрипторов молекулярных полей и числовым значением биологической активности при помощи метода частичных наименьших квадратов (Partial Least Squares — PLS) основаны методы 3D-QSAR, наиболее известным из которых является CoMFA[9].
6. Константы заместителей впервые были введены Л. П. Гамметом в рамках уравнения, получившего его имя, которое связывает константы скорости реакции сконстантами равновесия для некоторых классов органических реакций. Константы заместителей вошли в практику QSAR после появления уравнения Ганча-Фуджиты, связывающего биологическую активность с константами заместителей и значением липофильности. В настоящее время известно несколько десятков констант заместителей.
7. Фармакофорные дескрипторы показывают, могут ли простейшие фармакофоры, состоящие из пар или троек фармакофорных центров со специфицированным расстоянием между ними, содержатся внутри анализируемой молекулы.
8. Дескрипторы молекулярного подобия указывают на меру сходства (молекулярного подобия) с соединениями из обучающей выборки.
Индекс Винера (англ. Wiener index), известный также как число Винера (англ. Wiener number), — топологический индекс неориентированного графа 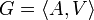 , определяемый как сумма кратчайших путей (англ.) d(vi,vj) между вершинами графа:
, определяемый как сумма кратчайших путей (англ.) d(vi,vj) между вершинами графа:
. 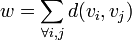 .
.
Индекс Рандича ( англ. Randić index), известный также как индекс связности неориентированного графа , является суммой вкладов по ребрам 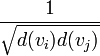 , где vi и vj — вершины, образующие ребро, d (vk) — степень вершины vk:
, где vi и vj — вершины, образующие ребро, d (vk) — степень вершины vk:
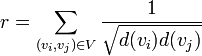 .
.
Индекс Рандича характеризуется неплохой дифференцирующей способностью, однако не является полным инвариантом. Для приведенных ниже пар графов он совпадает, хотя графы не являются изоморфными.
| Параметр | ||
| Граф | 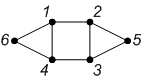 | 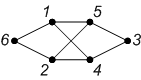 |
| Мини-код μ min | ||
| Индекс Рандича r | 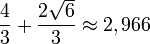 | |
| Параметр | ||
| Граф | 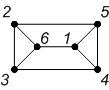 | 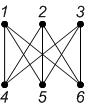 |
| Мини-код μ min | ||
| Индекс Рандича r |
Моделирование свойств при невекторном описании химических соединений осуществляется либо при помощи нейронных сетей специальных архитектур, позволяющих работать непосредственно с матрицами смежности молекулярных графов, либо при помощи ядерных (kernel) методов с использованием специальных графовых (либо химических, фармакофорных) ядер.
Примерами служащих для этой цели нейронных сетей со специальной архитектурой являются:
1. BPZ
2. ChemNet
3. CCS
4. MolNet
5. Graph machines
Примерами служащих для этой цели графовых (либо химических, фармакофорных) ядер являются:
1. Marginalized graph kernel
2. Optimal assignment kernel
3. Pharmacophore kernel
Вопрос №6. Понятие о статистических методах, применяемых в QSAR. Множественная линейная регрессия, пошаговая регрессия. Понятие о статистических критериях. Методы скользящего контроля, разбиение выборки на обучающую и контрольную.
Ответ на этот вопрос подробно рассмотрен в файле формата MS Word “Statistics_in_QSAR_1”, а также в презентации.
Вопрос №9. Понятие об обратной задаче в QSAR. Реконструкция структур по топологическим индексам, решение задачи на примере индекса Рандича. Генераторы химических структур, типы генераторов и области их применения. Генерация структур из фрагментов для целей QSAR. Роль структурных ограничений в генерации, понятие о комбинаторном взрыве. Отбор оптимальных структур с помощью моделей «структура-активность».
Обратная задача в проблеме «структура-свойство»(текст от лица Баскина, надеюсь, вы знаете, кто это такой)
В 1989 г. нами впервые была сформулирована т. н. обратная задача в проблеме «структура-свойство», суть которой заключается в поиске структур химических соединений, обладающих свойствами, лежащими в пределах заранее заданных интервалов [1]. Эта задача была нами решена для некоторых примеров путем ее сведения к комбинаторной задаче из области дискретной математики по перечислению графов, значения определенных инвариантов которых лежат в заданных интервалах [2][3][4] (см. обзор[5]). С тех пор понятие об обратной задаче в проблеме «структура-свойство» прочно вошло в область хемоинформатики, а первая наша статья на эту тему на английском языке до сих пор является самой цитируемой из моих публикаций[6].
В настоящий момент мною идет переосмысление этой задачи и в дальнейшем планируется ее переформулировать как задачу построения генеративных статистических моделей, позволяющих по заданному набору свойств найти условное статистическое распределение графов, процедуру сэмплинга из которого можно рассматривать как стохастический генератор химических структур, обладающих заранее заданным набором свойств. До самого последнего времени построение подобных моделей не представлялось реальным ввиду практически полной неразвитости математического аппарата построения генеративных статистических моделей графов, однако за последние несколько лет был опубликован ряд важных работ в этом направлении, которые позволяют взглянуть на эту область с определенным оптимизмом.
В методологии QSAR выделяют прямую и обратную задачи. Прямая задача QSAR заключается в предсказании активности на основания знания структуры. Обратной задачей QSAR является конструирование химических структур с заданными величинами активностей.
Поскольку речь идет о биологической активности, понятию QSAR близко понятие компьютерное моделирование лекарственных препаратов (или компьютерный дизайн лекарств), в англоязычной литературе термин более устоялся - Computer Aided Drug Design, или сокращенно CADD - савокупность вычислительных методов (в том числе и методов QSAR) и программ, используемых для направленного молекулярного дизайна лекарств (более корректно было бы говорить о потенциальных лекарствах или же о соединениях-лидерах). Хотя о чисто вычислительном дизайне говорить еще рано, поскольку многие свойства потенциальных лекарств в настоящее время можно определить исключительно экспериментальным путем (вычислительные же оценки носят качественный характер). CADD (компьютерный дизайн лекарств) можно рассматривать частным (хотя и наиболее изучаемым) направлением CAMD (Computer Aided Molecular Design), компьютерного молекулярного дизайна. Таким образом компьютерный молекулярный дизайн представляет собой савокупность подходов (методов, программ), используемых для молекулярного моделирования. Фактически экспоненциальное развитие вычислительных методов в последнее десятилетие связано главным образом с ростом мощностей вычислительных ресурсов и во вторую очередь - с развитием разных методов и подходов.
Комбинаторный взрыв — термин, используемый для описания эффекта резкого («взрывного») роста временной сложности алгоритма при увеличении размера входных данных задачи.
Более точно это означает, что рассматриваемый алгоритм не является полиномиальным, то есть время решения задачи не ограничено никаким многочленом от длины входа. Обычно такие задачи имеют экспоненциальную или даже сверхэкспоненциальную сложность.
Происхождение названия связано с тем, что для решения задачи не удается найти иного способа[источник не указан 561 день], кроме полного перебора всех возможных вариантов. В этом случае время, требуемое для решения, пропорционально количеству всех возможных конфигураций, которое определяется из тех или иных комбинаторных соображений (сочетания, перестановки).
Для обхода проблемы комбинаторного взрыва ищут специальные методы решения, в частности, применяют эвристические алгоритмы.