 2017-12-16
2017-12-16 1477
1477РАЗДЕЛ II. Общие закономерности химических процессов.
ТЕМА 8. Химическая кинетика и катализ
§1. Скорость химической реакции, ее зависимость от
различных факторов. Реакции I-го и II-го порядка
Законы термодинамики позволяют определить потенциальные возможности любого химического процесса, но ничего не говорят о его природе, характере, механизме и скорости.
Раздел химии, изучающий количественные закономерности и механизмы химических превращений, называют химической кинетикой.
Главное предназначение кинетики - обеспечение возможности управлять процессами, создавать продукты с заданными свойствами, предотвращать нежелательные побочные процессы.
Наряду со свободной энергией (ΔG и ΔF) количественной характеристикой потенциальных возможностей химического процесса является его скорость.
Скоростью химического процесса называют изменение количества вещества реакционной системы в единицу времени в единице объема реакционного пространства. Таким образом определенная скорость обозначается буквой υ или с и называется средней скоростью процесса за определенный промежуток времени.
В зависимости от природы химического процесса меняется и характер реакционного пространства. Так, если химический процесс совершается в гомогенной среде (и реагенты и продукты находятся в одном агрегатном состоянии), реакционным пространством для него служит объем сосуда, заполненный реагентами. В этом случае:
Средняя скорость гомогенной химической реакции определяется по изменению концентрации любого из реагентов или продуктов в
единицу времени; υi = ±ΔСi/Δτ. На рис.52 представлено изменение концентрации реагентов и продуктов во время совершения химической реакции.
Как видим, ΔСiреаг= Сi2 – Сi1<0, а ΔСiпрод = Сi2 – Ci1>0, скорость же процесса по определению не может быть отрицательной величиной, вот почему при мониторинге скорости по реагентам перед дробью ставится знак
минус «–».
Однако, средняя скорость не позволяет достаточно быстро реагировать на те или иные изменения во время химического процесса, особенно если эта
скорость высока.
 |
Рис.52. Изменение концентрации реагентов и продуктов реакции
за определенный промежуток времени.
Для мониторинга непрерывных процессов необходимо знать величину скорости в любой конкретный момент времени, т.е. определять мгновенную или истинную скорость процесса, а не среднюю.
Истинная скорость химического процесса характеризуется изменением концентрации любого компонента химической реакции (продукта или реагента) в данный конкретный момент времени.
С математической точки зрения это означает, что истинная скорость химического процесса по компоненту i (υi) будет рассчитываться как предел средней скорости при Δτ→0, т.е. υi = lim(±ΔСi/Δτ) = ±dСi/dτ.
Уравнение υi = ±dСi/dτ или υ = ±dС/dτ называется кинетическим уравнением для любого химического процесса. Скорость как физическая величина измеряется в [моль/л·с].
На скорость химического процесса влияет очень много различных факторов: природа реагентов и характер взаимодействия между ними (одни реакции идут мгновенно и со взрывом, а другие медленно, практически незаметно и тянутся десятками и сотнями лет), концентрация вступающих в реакцию исходных веществ, температура процесса, внешнее воздействие (например, излучение или пластическая деформация), присутствие катализатора, и даже иногда форма сосуда, в котором совершается реакция.
Влияние концентрации участников реакции на скорость процесса определяется экспериментальным путем. Взаимодействие между частицами реагентов происходит лишь в момент их реального физического столкновения, причем, с силой, достаточной для перехода кинетической энергии движущихся частиц в потенциальную энергию взаимодействия между ними. Если температура реакционной среды не изменяется, то число столкновений между частицами будет зависеть от количества самих частиц (чем их больше, тем больше вероятность эффективных соударений между ними). Следовательно, увеличение концентрации компонентов системы увеличивает скорость химического процесса.
Эта зависимость была установлена эмпирическим путем в 1864-1867гг норвежскими химиками К. Гульдбергом и П. Вааге и носит название основного постулата химической кинетики или закона действующих масс для кинетики.
Прежде чем мы сформулируем этот закон, запишем в виде схемы в общем виде уравнение реакции практически необратимого химического процесса (теоретически, как мы помним, все процессы являются обратимыми):
аА + bВ + dD + … = mM + nN + ℓL + … В этом уравнении обозначим молярные концентрации реагентов, возведенные в степени, соответствующие коэффициентам, с которыми эти реагенты входят в уравнение химической реакции, как Сμа(А); Сμb(В); Сμd(D). Такие величины в кинетике называют действующими массами реагентов.
Теперь сформулируем сам закон действующих масс:
Скорость практически необратимой химической реакции находится в прямой пропорциональной зависимости от действующих масс ее реагентов.
Математическое выражение закона действующих масс имеет вид:
υ = k·Сμа(А)·Сμb(В)·Сμd(D)…
Коэффициент пропорциональности (k) в законе действующих масс имеет физический смысл и называется константой скорости химической реакции. Легко установить, что при Сμ = 1моль/л скорость процесса υ = k, следовательно, «k» не зависит от концентрации реагентов, но зависит от их природы и от температуры, поэтому константа скорости является главной характеристикой любого химического процесса.
Закон действующих масс в представленном виде носит обобщенный характер. Показатели степени, в которые возведены концентрации реагентов (a,b,d…) называют «порядком реакции по реагенту A, B, D …», т. е.:
a = nA – порядок реакции по реагенту А;
b = nB – порядок реакции по реагенту В;
d = nD – порядок реакции по реагенту D и т.д.
Сумма порядков реакции по реагентам определяет общий порядок конкретной химической реакции (n): n = nA + nB + nD + …= ∑ni, где ni – порядок реакции по i – тому компоненту.
Например, для реакции 2СО + О2 = 2СО2 скорость химической реакции (υ), согласно закону действующих масс, выразится следующим уравнением: υ = k·Сμ2(СО)·Сμ(О2), а порядок этой реакции (n) будет равен n=2+1=3. Если процесс идет при эквивалентных соотношениях реагентов и Сμ (СО) = Сμ (О2), то кинетическое уравнение примет вид
υ = kСμ3, что прямо указывает на его порядок.
Для реакции N2 + O2 = 2NO скорость определится как
υ = k·Сμ(N2)·Cμ(O2) или υ = К·Сμ2, что указывает на 2-й порядок. А вот реакция разложения кислорода на атомарный кислород О2 = 2О˙ является реакцией первого порядка. Уравнение υ = f (Cn) называют уравнением формальной кинетики и по нему эмпирическим путем определяют скорость для реакций 1-го, 2-го, …, n –го порядка.
Так, скорость реакции 1-го порядка подчиняется уравнению υ=kСμ (по закону действующих масс) и уравнению υ= – dCμ/dτ (по определению понятия скорости). Приравнивая эти два уравнения, получим: dCμ/dτ = – kСμ
или, после разделения переменных, dCμ/Сμ = – kdτ. Решение полученного дифференциального уравнения при первоначальных условиях, что τ0=0 и Сμ при τ0 равна Сμ=C0, приведет к выражению: ∫dCμ/Сμ =∫ – kdτ, а далее при
взятии интеграла Сμ=C0·e-kτ. Подставив полученное значение концентрации реагента Сμ в закон действующих масс, получим окончательное выражение для скорости реакции 1-го порядка: υІ=kІ ·C0 ·e–kτ.
Если дифференциальное уравнение решать относительно константы скорости реакции k, то после его логарифмирования получим
 ℓnCμ = ℓnCo – kτ и, следовательно, kI = τ-1·ℓnCo/Cμ. На рис. 53 показан график зависимости ℓnCμ от τ, представляющий собой прямую линию. Отрезок, отсекаемый прямой на оси ординат, равен ℓnCo, а тангенс угла наклона прямой к оси абсцисс дает возможность определить константу скорости реакции первого порядка kI = tgα.
ℓnCμ = ℓnCo – kτ и, следовательно, kI = τ-1·ℓnCo/Cμ. На рис. 53 показан график зависимости ℓnCμ от τ, представляющий собой прямую линию. Отрезок, отсекаемый прямой на оси ординат, равен ℓnCo, а тангенс угла наклона прямой к оси абсцисс дает возможность определить константу скорости реакции первого порядка kI = tgα.
Большинство ядерных реакций являются реакциями 1-го порядка и для них характерно использование параметра «период полураспада τ½» - это время, в течение которого концентрация реагента уменьшается вдвое по сравнению с начальной концентрацией.
Для расчета τ½ в выражение для константы скорости реакции первого порядка подставим вместо τ – τ½, а вместо Сμ – Со/2. В результате получим:
kI = (1/τ½)·ℓn2, или τ½I= ℓn2/kI = 0,693/kI.
Как видим, период полураспада для реакций I-го порядка не зависит от концентрации.
Скорость реакции 2-го порядка для двухкомпонентной системы А + В = АВ подчиняется кинетическому уравнению υІІ=kІІ·Cμ(A)·Cμ(B). Если принять, что в этом уравнении Cμ(A) = Cμ(B), то υІІ=kІІ·Cμ2. В то же время, согласно основному постулату химической кинетики υ= -dCμ/dτ.
Объединим оба уравнения для скорости реакции в систему и, разделив переменные, получим дифференциальное уравнение dCμ/ Cμ2 = –kІІ·dτ. Интегрируем его при условии, что время изменяется от τ0=0 до τ, а концентрация – от C0 до Cμ: ∫dCμ/ Cμ2 =∫ –kІІ·dτ. При решении получим Cμ=C0/(1+kII·Co·τ). Это значение концентрации реагентов подставим в уравнение для скорости реакции 2-го порядка и решим его относительно υІІ и относительно kІІ. В результате получим уравнения, с помощью которых можно произвести расчеты скорости и константы скорости реакции 2-го порядка:
Cо2 1 1 1 (Со-Сμ)
υІІ=kІІ·——————; kІІ= —·(— – —)=-------------- ·
(1+kІІ·Cо·τ)2 τ Cμ Cо τCμCо
 |
Рис.53. Логарифмическая зависимость концентрации реагента
от времени протекания реакции 1-го порядка.
 Если построить график зависимости концентрации реагента от времени реакции 2-го порядка в координатах (1/Сμ ÷τ), то он также будет представлять из себя прямую линию, отсекающую на оси ординат отрезок, равный 1/С0, а по тангенсу угла наклона прямой к оси абсцисс можно определить константу скорости реакции II-го порядка: k = tgα (см. рис. 54).
Если построить график зависимости концентрации реагента от времени реакции 2-го порядка в координатах (1/Сμ ÷τ), то он также будет представлять из себя прямую линию, отсекающую на оси ординат отрезок, равный 1/С0, а по тангенсу угла наклона прямой к оси абсцисс можно определить константу скорости реакции II-го порядка: k = tgα (см. рис. 54).
 Период полураспада для реакций второго порядка определяется по уравнению: τ½ІІ=1/kІІ·C0.
Период полураспада для реакций второго порядка определяется по уравнению: τ½ІІ=1/kІІ·C0.
Все приведенные нами кинетические уравнения относились к гомогенным системам. Гетерогенные реакции протекают на поверхности раздела фаз, именно поверхность и служит реакционным пространством в этих случаях. Тогда закон действующих масс будет звучать несколько иначе:
Скорость гетерогенной химической реакции находится в прямой пропорциональной зависимости от концентрации (действующей массы) неконденсированной (наименее конденсированной) фазы реагентов и площади реакционной поверхности (площади поверхности
конденсированной фазы).
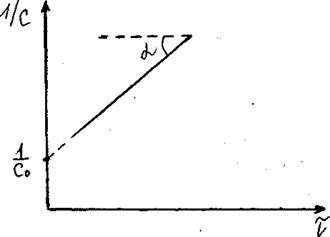 Рис.54. Зависимость скорости реакции второго порядка от времени процесса.
Рис.54. Зависимость скорости реакции второго порядка от времени процесса.
Следовательно, для гетерогенной реакции I-го порядка выражение скорости будет иметь вид: υІ=kІ·Cμ·S, где Сμ – концентрация газообразного реагента, S – площадь поверхности твердого реагента.
Например, скорость гетерогенной реакции СаО(т) + СО2(г) = СаСО3(т) определится по формуле υІ=kІ·Cμ(СО2)·S(СаО). Как следует из приведенной формулы, чем больше площадь поверхности твердого реагента, тем выше скорость гетерогенной реакции. Вот почему требуется измельчение твердой фазы для ускорения процесса, либо наращивание пористого покрытия, увеличивающего количество активных центров реакционного пространства.
Если реакция происходит между твердыми веществами, то ее скорость вообще не будет зависеть от концентрации реагентов, а только лишь от развитости поверхности взаимодействующих компонентов. Такие реакции имеют нулевой порядок по всем реагентам и для них υ0=υ0–k·τ, то есть, скорость реакции линейно уменьшается во времени.
С другой стороны, в гетерогенных процессах с участием жидких и твердых реагентов на их скорость оказывает влияние интенсивность доставки реагентов в реакционное пространство. Именно поэтому в выражении для скорости используется не объемная концентрация вещества Сμ(раствора), а молярная концентрация на поверхности твердой фазы в зоне реакции Сμ(S), ее принято еще обозначать символом Сs. С учетом сказанного, уравнение скорости гетерогенной реакции первого порядка примет вид:
υІ = kІ·Cs·S.
Перемешивание увеличивает скорость доставки реагентов и выравнивает концентрацию на поверхности реакционной зоны, поэтому гетерогенные процессы проводят при постоянном перемешивании.
Влияние температуры на реакционный процесс также оказывается достаточно сложным. Повышение температуры ускоряет большинство химических реакций, т.к. при этом увеличивается кинетическая энергия частиц, участвующих во взаимодействии, и возрастает скорость их перемещения в реакционном пространстве.
Согласно эмпирическому правилу Вант-Гоффа (1878г):
повышение температуры на каждые 100 увеличивает скорость химической реакции в 2÷4 раза.
Уравнение Вант-Гоффа имеет вид:
(Т2-Т1)/ 10
υT2/υT1 = γ. В этом уравнении υT2 и υT1 – скорости реакции при температурах Т2 и Т1, γ – температурный коэффициент Вант-Гоффа, принимающий, по определению, значения в пределах γ = 2÷4.
Поскольку скорости и константы скоростей химической реакции пропорциональны друг другу, а время протекания реакции обратно пропорционально ее скорости, то уравнение Вант-Гоффа относительно констант и времени протекания реакции при различных температурах можно записать следующим образом:
Т 2 -Т 1 Это уравнение можно использовать лишь для 10 расчетов в узком интервале температур, близких
kT2 ‗ τ1 ‗ γ к 100 0 С. Установлено, что температурный
kT1 τ2 коэффициент Вант - Гоффа для эндотермических
реакций выше, чем для экзотермических: γэнд>γэкз.
Более точным уравнением, показывающим взаимосвязь между константой скорости химического процесса и температурой, и действующим в неограниченном диапазоне температур, является уравнение С. Аррениуса (1889г, Швеция): k = A·e-Ea/RT. В этом уравнении: k – константа скорости химической реакции при температуре Т; А – стерический множитель, показывающий долю наиболее эффективных (приводящих к взаимодействию) столкновений между реагентами; Еа – энергия активации реагентов, зависящая от их природы (это та избыточная энергия, которой должны обладать частицы реагирующих веществ, чтобы между ними произошло взаимодействие). Для большинства химических реакций энергия активации Еа находится в пределах Еа = 40 ÷ 400 кДж/моль.
Как и все предыдущие кинетические уравнения, уравнение Аррениуса можно преобразовать в логарифмическую форму. Тогда оно приобретет вид:
ℓnk = ℓnA–Ea/RT. Как следует из уравнения, в координатах ℓnk ÷ 1/Т
график зависимости константы скорости от температуры представляет собой прямую линию, отсекающую на ординате отрезок, равный lnA, а тангенс угла наклона прямой к оси абсцисс позволяет определить энергию активации: tgα=–Ea/R (см. рис. 55).



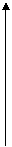 ℓnk
ℓnk
 | |||||
 | |||||
 | |||||

 ℓnA
ℓnA
 |







 1/T
1/T
Рис.55. Зависимость константы скорости реакции от температуры
Это уравнение используется при расчете постоянных для заданных химических реагентов величин А и Еа, и для мониторинга зависимости скорости химической реакции от температуры.
§2. Механизмы химических превращений. Простые и сложные
реакции. Цепные и фотохимические процессы. Теория
активированного комплекса С. Аррениуса
Уравнения формальной кинетики υ = f(Cμ) и υ = f(T) позволяют рассчитать скорости реакций в зависимости от условий их протекания, но не дают точных представлений об их механизмах. Однако выявление механизмов химических превращений является важнейшей фундаментальной задачей химии, т.к. позволяет управлять химическим процессом.
Все химические реакции подразделяются на простые и сложные.
Простые реакции протекают в один этап или в одну стадию (из реагентов сразу без побочных явлений получаются продукты реакции). Их называют одностадийными или элементарными.
Сложные реакции реализуются через несколько стадий и протекают последовательно или параллельно, или последовательно - параллельно.
Механизмом химического процесса называют полную последовательность всех его элементарных стадий.
Согласно теории вероятности, в каждой стадии химической реакции может участвовать одна, две, реже три частицы (вероятность эффективного столкновения в конкретной точке реакционного пространства в конкретный момент времени более трех частиц равна нулю).
Количество молекул реагентов, принимающих участие в простейшей (элементарной) стадии химического процесса, называется ее молекулярностью. Это количество может равняться одной частице (мономолекулярная стадия), двум частицам (бимолекулярная стадия), иногда трем частицам (тримолекулярная стадия). Реакции с молекулярностью больше трех неизвестны.
Если общий химический процесс состоит только из одной стадии, то его порядок совпадает с молекулярностью. И тогда моно-, би- и тримолекулярные стадии становятся соответственно реакциями 1, 2 или 3 порядка. Приведем примеры одностадийных химических процессов.
Н2 = 2Н˙; ²²688Ra = ²²²86Rn + 42He –мономолекулярные реакции 1-го порядка.
NO + O3 = NO2 + O2 – бимолекулярная реакция 2-го порядка.
2NO + H2 = N2O + H2O; 2NO + O2 = 2NO2 – тримолекулярные реакции 3-го
порядка.
Кинетические уравнения приведенных реакций полностью совпадают с законом действующих масс. Однако таких реакций в природе чрезвычайно мало. Большинство химических процессов являются сложными и многостадийными.
Например, простая, на первый взгляд, реакция разложения оксида азота (V), подчиняющаяся общему уравнению 2N2O5 = 4NO2 + O2, должна быть бимолекулярной реакцией II-го порядка. Однако экспериментальное исследование зависимости скорости реакции от концентрации реагента показывает, что у этой реакции первый порядок. Почему?
На самом деле процесс разложения оксида азота (V) протекает через три последовательные стадии:
(1) N2O5 = NO2 + NO3˙ – быстрая мономолекулярная стадия;
(2) NO3˙ = NO˙ + O2 – медленная мономолекулярная стадия;
(3) NO3˙ + NO˙ = 2NO2 – быстрая бимолекулярная стадия.
В кинетике, впрочем, как и в любом другом (нехимическом) процессе его скорость всегда определяется по самому медленному участку, в данном случае, по самой медленной стадии. Самую медленную стадию называют лимитирующей. Она, как правило, является необратимой (направлена в одну сторону ─►), что же касается быстрых стадий, то они относятся к обратимым химическим процессам (идут в прямом и обратном направлении ◄═►).
Изложенное позволяет нам кинетически грамотно записать реакцию разложения N2O5 в виде трех процессов, два из которых являются обратимыми, а один - необратимый лимитирующий:
(1) N2O5◄═► NO2 + NO3˙
(2) NO3˙ ─► NO˙ + O2
(3) NO3˙+ NO˙ ◄═► 2NO2.
Таким образом, лимитирующей стадией в данном процессе является вторая. Но мы видим, что она мономолекулярная и I-го порядка. На самом деле в первой и во второй стадиях этого сложного процесса образуются промежуточные короткоживущие частицы (радикалы NO3˙и NO˙). В кинетике их называют интермедиатами. При этом всегда выполняется условие, что количество образовавшихся и израсходованных интермедиатов должно быть одинаковым. А если это так, то скорость второй (медленной) стадии будет зависеть от исходной концентрации реагента N2O5, образующего интермедиат NO3˙ по первой стадии, для которой по закону действующих масс υ=kCμ(N2O5). Вот и получилось, что этот сложный процесс подчиняется уравнению реакции I-го порядка.
В приведенном примере реакция разложения N2O5 оказалась сложной последовательной трех стадийной реакцией I-го порядка мономолекулярной по лимитирующей стадии.
Параллельные сложные реакции протекают одновременно по нескольким направлениям. Схема такого процесса выглядит примерно так:
┌►D
А + В =┤
└►E
Например, при разложении гидразина N2H4 при одних и тех же условиях образуется два набора продуктов по параллельно идущим реакциям
N2H4 = N2 + 2H2 (1, быстрая) и 3N2H4 = 4NH3 + N2 (2, медленная).
В таких реакциях интермедиаты отсутствуют. Средняя скорость всего процесса будет зависеть от лимитирующей реакции (2) вследствие ее меньшей вероятности (тримолекулярный процесс). Порядок реакции в таких системах всегда будет дробным.
Если с одним и тем же реагентом одновременно взаимодействуют два или более веществ, то такие сложные процессы называются сопряженными реакциями. Самая простая схема сопряженных реакций выглядит как
{А + В = АВ и одновременно A + D = AD}.
Сопряженные реакции наиболее характерны для растворов. Например, при добавлении газообразного сероводорода H2S в раствор хлорида цинка ZnCl2 проходят две сопряженные реакции:
Zn2+ + H2S = ZnS↓ + 2H+;
Zn2+ + 4H2O = [Zn(OH)4]2- + 4H+.
В некоторых случаях сопряжение реакций может вызвать их ускорение.
Наиболее сложными являются последовательно – параллельные реакции, в которых продукты одного процесса становятся реагентами для последующих процессов, ускоряя каждую параллельную и последовательную реакцию по мере накопления интермедиатов.
Например, реакция окисления бромноватой кислоты (HBrO3) метамышьяковистой кислотой (НАsО2) является последовательно-параллельным процессом, состоящим из 4-х последовательных стадий, развивающихся при этом и параллельно:
HBrO3 + HAsO2 = H3AsO4 + HBr (медл.)
HBrO3 + HBr = HBrO + HBrO2 (быстр.)
HAsO2 + HBrO + H2O = HBr + H3AsO4 (быстр.)
2HAsO2 + HBrO2 + 2H2O = 2H3AsO4 + HBr (быстр.).
Многие биохимические процессы в живых организмах протекают по типу сопряженных реакций.
Колебательные реакции – это периодические процессы, характеризующиеся колебаниями концентраций некоторых промежуточных соединений и, соответственно, скоростей их превращений.
Например, при взаимодействии лимонной кислоты с бромноватым калием KBrO3 в присутствии трехзарядных катионов церия Се3+, раствор регулярно меняет окраску от бесцветной (Се – III) к желтой (Се-IV) и обратно. Эти явления объясняются принципами термодинамики необратимых процессов и выявляются только эмпирическим путем.
Цепные реакции. Некоторые химические реакции под влиянием внешних факторов (давление, температура, катализатор, форма сосуда) ускоряются лавинообразно. Впервые идея о цепном механизме подобных процессов была высказана русским ученым Н.А. Шиловым в 1905г, позднее за развитие теории цепных реакций академик Н.Н.Семенов был удостоен Нобелевской премии.
Цепные реакции начинаются с инициирования, т.е. образования радикалов (осколков молекул, атомов с не спаренными электронами), обладающих повышенной реакционной способностью, например, Cl˙,O˙,OH˙,H3C˙, HS˙ и т.п. Инициирование происходит под воздействием света, радиоактивного излучения, нагревания.
Появление радикалов в реакционной среде называют зарождением цепи. Например, реакция синтеза хлороводорода Н2 + Cl2 = 2HCl на свету приобретает характер цепной реакции. Ее механизм можно представить следующим образом:
а) инициирование реакции и зарождение цепи происходит под действием светового излучения, в молекуле хлора при этом разрывается ковалентная неполярная связь и образуется атомарный хлор-радикал, содержащий не спаренный электрон:
ħν
Cl:Cl → Cl˙(↓) + Cl˙(↑).
В этом уравнении стрелками (↑↓) показано направление спинов не спаренных электронов в радикалах хлора; ħν – энергия светового излучения, поглощаемая молекулой хлора для инициирования ее активности. Опыт показывает, что для образования 1 моля радикалов хлора вполне достаточно 8 кДж энергии. Таким образом, энергия активации данной цепной реакции Еа составляет Еа = 8 кДж/моль;
б) рост цепи. Надо заметить, что количество возбужденных молекул хлора, в которых произошел разрыв связей с образованием радикалов, остается незначительным по сравнению с общим количеством молекул. Однако этого вполне достаточно, чтобы высвободившиеся радикалы дальше вступили во взаимодействие с другими молекулами хлора и водорода и произошел рост цепи:
Cl˙ + H2 → HCl + H˙
H˙ + Cl2 → HCl + Cl˙ … и т.д. Как видим, радикалы воспроизводятся и цепная реакция продолжается дальше. Причем, для ее развития не требуется большого количества дополнительной энергии, вполне хватает энергии, поглощенной от светового излучения во время инициирования цепи;
в) обрыв цепи. Если радикалы Сl˙ и Н˙ встречаются в реакционном пространстве, то их взаимодействие приводит к понижению активности системы и к обрыву цепи:
Н˙ + Н˙ → Н2
Cl˙ + Cl˙ → Cl2
H˙ + Cl˙ → HCl. Обрыв цепи может произойти и на частицах примеси, загрязнений, выступах поверхности сосуда и т.п.
На данном примере мы рассмотрели механизм простой цепной реакции, однако возможны и более сложные процессы.
Разветвленные цепные реакции возникают в том случае, если вместо одного вида зарождаются сразу два и более видов радикалов. В этом случае цепь растет стремительно (см. рис.56), резко возрастает скорость всех процессов, что часто приводит к взрыву. Например, взрыв гремучего газа, состоящего из двух объемов водорода и одного объема кислорода, происходит по схеме: 2Н2+О2=2Н2О (суммарное уравнение)
а) зарождение цепи Н2 + О2 → 2НО˙;
б) рост цепи НО˙ + Н2 → Н ˙ + Н2О,
НО˙ + О2 → О˙ + Н2О;
в) разветвление цепи Н ˙ + О2 → НО˙ + О˙
О˙ + Н2 → Н˙ + НО˙ и т.д.
Возрастание радикальной активности системы приводит к неуправляемости процесса и к взрыву.
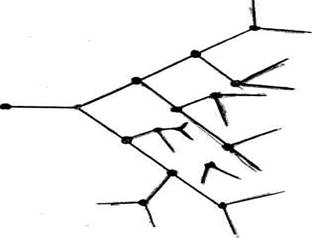 |
Рис.56. Схема разветвленной цепной реакции.
Цепные реакции встречаются достаточно часто (это и горение топлива в двигателях внутреннего сгорания, и взрывы в атмосфере, детонация природного газа в шахтах, реакции полимеризации, процессы радиоактивного распада и т.п.). Знание механизмов протекания процессов дает возможность предотвратить аварийные ситуации при промышленном применении взрывоопасных систем.
Фотохимические реакции. Таковыми называют процессы, возникающие в реакционной системе под действием поглощенной ею световой энергии (ħν). В отличие от цепных реакций, в данном случае не образуется радикалов, процесс идет по иному (менее активному механизму). Часто компонент системы, активированный световой энергией, выступает в роли катализатора, инициируя процесс и высвобождаясь при его окончании.
Например, при фотосинтезе вследствие поглощения видимого света зеленым красителем - хлорофиллом, содержащимся в листьях растений, последний выступает в роли катализатора взаимодействия углекислого газа и воды с образованием гликозидов и кислорода:
ħν
6СО2 + 6Н2О → С6Н12О6 + 6О2.
Тепловой эффект такого процесса достаточно велик и составляет ΔН = 2815 кДж/моль.
Для фотохимических процессов эмпирическим путем установлены два закона. Первый закон фотохимии (Т. Гротхгус) гласит:
Фотохимически активным является только поглощенный свет.
Второй закон фотохимии (А. Эйнштейн – Г. Штарк): каждый поглощенный фотон светового излучения вызывает первичное изменение только одной молекулы.
Количество молекул, активированных при поглощении одного кванта энергии (одного фотона ħν) и вступивших во взаимодействие с другими молекулами, называют квантовым выходом фотохимического процесса(η). Из второго закона фотохимии следует, что в этих процессах квантовый выход не может превышать единицу, но может быть и меньше, если активизировавшейся молекуле придется столкнуться с себе подобной или разрядиться о стенку сосуда. Таким образом, в фотохимических процессах
η ≤ 1 (для сравнения, квантовый выход в цепных реакциях η >100).
Фотохимические процессы широко используются в фотографии. Все помнят, что на покрытой галогенидами серебра (чаще всего используется бромид серебра AgBr) фотопленке под действием света происходит разложение соли
ħν
2AgBr → 2Ag + Br2.
Выделяющееся при этом серебро ровным слоем покрывает поверхность в местах засвечивания, повторяя контуры фотографируемого объекта.
Современная фотография использует не серебряные фотоматериалы, например, соединения железа Fe и хрома Cr с диазонием, некоторые полимеры. Под воздействием света происходят фотохромные процессы на фотопластинах или пленках, и цвет соединения изменяется в местах засвечивания.
Фотохромные материалы применяются в светофильтрах, в лазерных установках, в устройствах для регистрации, индикации и обработки оптической информации.
Фотохимические процессы играют чрезвычайно важную роль для поддержания жизни на Земле. Мы уже упомянули о фотосинтезе, благодаря которому в атмосфере естественным путем уменьшается доля диоксида углерода СО2 и увеличивается концентрация молекулярного кислорода О2.
Под действием света (коротковолнового ультрафиолетового излучения с длиной волны λ< 240 нм) в верхних слоях атмосферы (на высоте свыше 70км) происходит ионизация газов и образование плазмы:
N2 + ħν → N2+ + ē
NO + ħν → NO+ + ē
O2 + ħν → O2+ + ē.
Наличие ионизированного слоя обеспечивает распространение радиоволн на нашей планете. На высоте, превышающей 50км, ионизированная плазма формирует слой атомарных газов: N2+ + ē → 2N
O2+ + ē → 2О
NO+ + ē → N + O.
Ниже его формируется озоновый слой по реакции О2 + О → О3, который является защитой жизни на Земле, т.к. опускаясь в нижние слои атмосферы и поглощая тепловое ультрафиолетовое излучение, озон вновь синтезирует молекулярный кислород: О3 + ħν → О2 + О.
К сожалению, защитный озоновый слой становится все менее плотным, в нем образуются т.н. озоновые дыры. Интенсивное производство и эксплуатация химических материалов, сопровождающееся выбросом в атмосферу оксидов азота, углерода, серы, приводит к «сжиганию» озона:
О3 + NO2 → NO + 2O2;
O3 + CO2 → CO + 2O2;
O3 + SO2 → SO3 + O2;
и выпадению кислотных дождей: SO3 + H2O → H2SO4.
Вот почему охрана атмосферного слоя становится чрезвычайно важной глобальной проблемой для всех земных цивилизаций.
Теория активированного комплекса С. Аррениуса (1889). Анализируя влияние различных факторов на скорость химических процессов, мы установили ее высокую чувствительность к свету и температуре.
Так, согласно правилу Вант-Гоффа, при нагревании реакционной системы на 100 град. скорость процесса может возрасти в 59000 раз (при температурном коэффициенте скорости γ = 3). Но как показывает кинетическая теория газов, при их нагревании скорость движения молекул увеличивается пропорционально √Т, а значит, и число столкновений между молекулами должно возрасти в таком же порядке – не более чем в 10 раз при нагревании на 1000 (√100 = 10). В чем причина столь сильного влияния температуры на скорость процесса?
С другой стороны, поместив в сосуд объемом 1л смесь газов количеством 1моль (6,02·1023 молекул), мы создаем условия, при которых каждая молекула в течение 1с сталкивается с другими молекулами не менее чем 1010 раз! Если бы каждое из этих столкновений приводило к химическому взаимодействию, то все реакции заканчивались бы мгновенно. Мы знаем, что на самом деле это не так. Значит, не все столкновения являются эффективными. Большинство молекул, столкнувшись, разлетаются в разные стороны как упругие шары. Почему?
Исследуя кинетику химических процессов, С. Аррениус предположил, что эффективность взаимодействия между молекулами газов зависит от двух факторов – их потенциальной энергии и пространственной ориентации. Ученый предположил, что результативными для химического взаимодействия будут лишь столкновения между активными молекулами, обладающими энергией выше среднестатистической всего реакционного пространства. Зная строение атомов и природу химической связи, мы понимаем, что для осуществления элементарного акта реакции необходимо, чтобы электронные оболочки атомов реагентов, преодолевая взаимное отталкивание, вторглись одна в другую. Это вызовет разрыв старых химических связей и образование новых, т.е. произойдет собственно химическое превращение. Для разрыва связей как раз и нужна дополнительная энергия (ее Аррениус назвал энергией активации Еа), которой обладают не все молекулы. На рис. 57 представлен график зависимости энергии реакционной системы от хода реакции (в качестве второго параметра может быть выбрана концентрация или парциальное давление реагентов, или время). Кривая повышения полной энергии системы во время разрыва старых химических связей носит название «энергетического барьера».
Применив статистическую теорию Максвелла-Больцмана о распределении большого числа частиц (N→∞) по их энергиям (на рис.58 приведена кривая распределения), Аррениус рассчитал количество активных молекул (Nакт), обладающих повышенной энергией по сравнению с
остальными в данной системе (No): Nакт = No·е –Еа/RT.

Рис.57. Энергетический барьер
химической реакции.
Исследуя зависимость скорости реакции от энергии активации, Аррениус обратил внимание на то, что многие сложные (многостадийные) процессы на практике требуют меньше энергии, чем показывает расчет.
 |
Рис.58. Кривая распределения N структурных единиц системы по энергиям
или кривая Максвелла-Больцмана.
Например, реакция синтеза иодида водорода по уравнению
Н2 + J2 = 2НJ
согласно расчетам, требует при нормальных условиях дополнительной энергии для разрыва химических связей в молекулах йода и водорода 571 кДж/моль. Однако тепловой эффект этой реакции оказался равным лишь 168 кДж/моль. Ученый предположил, что на самом деле в процессе синтеза система проходит через промежуточные стадии, образуя некие переходные состояния (активированные комплексы), когда прежние связи еще не разорваны, но лишь ослаблены, а новые еще не возникли, но уже наметились.
В приведенном примере ход реакции образования HJ можно представить следующей схемой:
Н – Н ┌H … H┐ Н Н
+ ──► │׃ ׃ │ ──► │ + │
J – J └J … J ┘ J J
Реагенты активированный комплекс продукты.
Время существования активированных комплексов невелико и составляет всего 10-14 – 10-11с. Но для образования такого переходного состояния требуются значительно меньшие запасы энергии, чем для полного разрыва связей в исходных молекулах. Как следует из приведенного механизма, энтальпийный фактор (энергия активации) существенно влияет на скорость химического процесса.
Вместе с тем, даже при соударении активных молекул реакция может не произойти, если молекулы не будут выгодно ориентированы в пространстве относительно друг друга. Убедимся в этом на том же примере синтеза HJ. Допустим, переходный комплекс имеет иное пространственное строение, и реакция совершается по радикальному механизму:
Н – Н + J – J ──► [ Н …H…J…J] ──► [H…H…J]˙ + J˙
Реагенты активированный комплекс интермедиаты.
Дальнейшее развитие цепи возможно лишь в том случае, если радикал J˙ окажется ориентированным относительно интермедиата [H…H…J]˙ со стороны водородной цепочки, тогда:
J˙ + [H…H…J]˙ ──► [J˙…H…H…J]˙ ──► H–J + H–J.
Интермедиаты активированный комплекс-II продукты.
Если же радикал J˙ будет сориентирован со стороны йодной цепочки интермедиата [H…H…J]˙, то произойдет обрыв цепи и система вернется в исходное состояние:
[H…H…J]˙ + J˙ ──► [H…H…J˙…J˙] ──► H–H + J–J.
Интермедиаты активированный комплекс-III реагенты.
Как следует из данной схемы, скорость химического процесса сильно зависит от пространственной ориентации молекул (энтропийный фактор).
В уравнении Аррениуса для константы скорости (k) любой химической реакции нашли свое отражение оба фактора (энтальпийный Еа и энтропийный А): ℓnk = ℓnA – Еа/RT.
Чем выше температура в реакционной среде, тем большее количество молекул будут обладать энергией активации, но тем менее упорядоченной становится система, в ней нарушается ориентация и резко снижается энтропийный фактор. Вот почему большинство реакций идут все же с малыми скоростями и являются обратимыми.

