2020-01-14
2020-01-14 211
211Выполнил: студент
2-го курса магистратуры
В.В. Новиков
Проверил:
Проф.. д.х.н. Ю.Г. Папулов
Тверь 2002
План
ВВЕДЕНИЕ
1. ОСНОВНЫЕ ПОДХОДЫ К МАТЕМАТИЧЕСКОМУ МОДЕЛИРОВАНИЮ МАКРОМОЛЕКУЛ
2. МЕХАНИЧЕСКАЯ МОДЕЛЬ МОЛЕКУЛЫ
3. МЕТОДЫ МОЛЕКУЛЯРНОЙ ДИНАМИКИ (МД)
4. МЕТОД МОНТЕ-КАРЛО (МК)
5. ОСОБЕННОСТИ КОМПЬЮТЕРНОГО ЭКСПЕРИМЕНТА
6. Применения компьютерного эксперимента
7. Преимущества компьютерного моделирования
СПИСОК ЛИТЕРАТУРЫ
Введение
В настоящее время полимеры имеют огромное значение для жизнедеятельности человека, но еще большую роль не только для жизни людей, но и жизни в целом, играют биологические макромолекулы, своеобразные природные полимеры: белки, нуклеиновые кислоты, полисахариды. В живой клетке все эти макромолекулы играют разные роли: хранение генетической информации, запас питательных веществ и энергии, самовоспроизводство клети и ее органелл, защита от проникновения инородных тел, транспорт различных молекул или ионов и многие другие, без которых невозможно представить жизнедеятельность клетки. Все биологические макромолекулы различны и по молекулярной массе, и по "внешнему" виду макромолекулы, то есть по конформации (от сфер до жестких палочек). Однако есть у них и общие черты:
1). Так как макромолекулы состоят из огромного (сотни тысяч - миллионы) числа атомов, то даже одна молекула в ряде случаев может рассматриваться как "молекулярная система".
2). Макромолекулы (белки или нуклеиновые кислоты) могут изменять свою конформацию в широких пределах, что очень сильно влияет на физические и физиологические свойства молекул (вязкость растворов, биологическая активность и т. п.).
3). Атомы в составе макромолекулы соединены ковалентной связью, в строгом порядке и представляют собой одно целое, кроме того, макромолекулы даже в относительно разбавленных растворах не могут двигаться независимо друг от друга. Поэтому по сравнению с такими же по химическому составу низкомолекулярными соединениями они имеют аномально низкую энтропию и, как следствие, высокую чувствительность к различным низкоэнергетическим воздействиям.
Изучение связи этих свойств со строением макромолекул и основными физическими факторами (температура, растворитель и др.) и составляет основу теоретических разделов науки о полимерах.
Развитие учения о полимерах можно разделить на несколько этапов. Первый этап, охватывает собой все достижения сделанные в этой области до конца 60-х годов XX века. Он завершился созданием конформационной статистики макромолекул, основы которой изложены в трех классических монографиях. Завершением этого этапа следует признать вручение Нобелевской премии по химии американскому физикохимику, профессору Стенфордского университета Полу Флори в 1974 году. Его исследования в этой области открыли дорогу методам конформационной статистики, связав свойства растворов полимеров с конформацией отдельных макромолекул в нем. Его теоретическое рассмотрение основывалось на приближенных феноменологических представлениях, что и было характерно для того этапа развития.
Следующий этап начался в 70-е годы и длится по сей день. Он ознаменован вхождением в науку о полимерах новых представлений, таких как идеи и методы флуктуационной теории фазовых переходов и физики твердого тела. Огромный вклад был сделан такими выдающимися физиками-теоретиками как француз П. - Ж де Жен и наш соотечественник И. М. Лившица. Де Жен, в частности, создал изящную и наглядную методологию описания равновесных и динамических свойств полимеров, названной теорией скейлинга. За это де Жен получил Нобелевскую премию по физике. В предисловии к своей монографии он называет три главных обстоятельства, сделавших возможным прогресс науки о полимерах: во-первых, внедрение мощных экспериментальных методов исследования, как метод рассеяния нейтронов, позволяющих экспериментально определить многие характеристики полимерных материалов, вплоть до конформации макромолекул и строения доменов. Во-вторых, привлечение существующих теоретических концепций из других разделов физики и создание новых теорий. И, наконец, в-третьих, интенсивное использование специальных методов машинного моделирования.
В последнее время роль вычислительной машины в науке сильно изменилась. Машина уже не только хранит и перерабатывает заложенные в нее экспериментальные данные, но и выступает в роли самостоятельного источника новых физических знаний. За этим новым и довольно неожиданным применением компьютеров утвердилось название компьютерный эксперимент. Основан он на математическом моделировании, которое применяется уже около двух веков, но только после появления электронно-вычислительных машин он стал применяться для изучения простых жидкостей и, именно, с его помощью были получены важные сведения о структуре и динамике конденсированных систем (в частности о структуре жидкой воды). Моделирование более сложных молекул полимеров и даже объектов живой природы начало развиваться позднее.
Роль компьютерного моделирования в методологии науки о полимерах сложно недооценить - оно произвело настоящую революцию в науке. До этого все естественные науки имели довольно строгое разделение на теоретическое и экспериментальное направления. Компьютерное моделирование, являясь, по сути, и тем и другим, сгладило это историческое деление. Со времени своего возникновения и по сей день, в этой области естествознания идут процессы "структурообразования": накапливаются методы, приемы моделирования, появляются новые (более точные) модели и т. д. С увеличением производительности компьютеров расширяется круг задач посильных компьютерному эксперименту. Так в настоящее время моделируется не структура чистого растворителя, а ассоциаты возникающие в растворах амфолитов, моделируются процессы проходящие в биологических мембранах и даже целые биологические реакции как, например фотосинтез.
Основные подходы к математическому моделированию макромолекул
Основная задача статистической теории - вычисление средних значений различных величин, которые характеризуют поведение системы в состоянии равновесия. Существуют два подхода к решению этой общей задачи. В первом случае среднее значение <А> некоторого свойства A (r, v),которое предполагается зависящим от совокупности координат- {r} и скоростей {v} частиц, определяют путем усреднения множества "мгновенных" значений A [r(t),v(t)], наблюдаемых в последовательные моменты времени t на достаточно протяженном интервале t:
А = 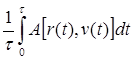 (1.1)
(1.1)
Этот подход, называемый усреднением по времени, исходит из того, что нам известны законы движения частиц системы.
Альтернативный путь вычисления средних значений параметров системы был намечен еще Больцманом, а затем развит Гиббсом в стройную теорию. Идея этого подхода заключается в том, что наблюдаемое свойство рассматривается не как среднее по времени, а как среднее по множеству различных состояний системы, которые возникают с определенной вероятностью. Такой подход называют усреднением по ансамблю. Вероятность (или частота) возникновения того или иного состояния пропорциональна его статистическому весу w=e- U /kT, где U - потенциальная энергия данной конфигурации, k - константа Больцмана, Т - абсолютная температура. В этом случае наблюдаемые средние значения даются общим выражением
А =∫ … ∫ A (r)w(r)dr / ∫ … ∫w(r)dr (1.2)
Оба фундаментальных принципа определения средних значений могут быть положены в основу вычислительных схем, реализуемых на компьютере. При этом необходимо знать лишь способ расчета потенциальной энергии системы как функции координат r. Результаты расчетов, какого - либо свойства одной системы вычисляемые по одному и другому пути должны совпадать при длительном времени наблюдения за системой в первом подходе и при очень большом числе испытаний во втором подходе.