 2015-03-27
2015-03-27 1764
1764Обнаруженные в предыдущем разделе сложные зависимости θ – рН и обсуждавшиеся в связи с этим факторы, влияющие на форму этой зависимости, требуют дальнейших исследований с привлечением других методов. Одним из наиболее продуктивных методов изучения энергетики взаимодействия, как известно, является калориметрия. Взаимодействие поверхности твердого тела с жидкостями изучается с помощью теплот смачивания.
IV.1. Некоторые литературные данные по исследованию
теплот смачивания
По определению [15], теплота смачивания равна разности энтальпий границ раздела твердое тело – жидкость и твердое тело – вакуум. Теплоты смачивания обычно невелики и успехи в их изучении были достигнуты; относительно недавно [15], после изобретения прецизионных микрокалориметров [307].
Теплота смачивания (Q) обычно положительна и в зависимости от природы оксида изменяется в пределах от 0,1 до 1,0 Дж/м2 [273,308-310]. Согласно литературным данным, теплоты смачивания кварца и силикатов довольно разнообразны. В работе [32] приводятся значения Q различных силикагелей и кварца порядка 0,10-0,25 Дж/м2. В [310] наряду с величинами того же порядка для аморфного SiO2 и аэросила приводится для кристаллического кварца величина 0,85 Дж/м2. Это значение Q, по нашему мнению, сильно завышено из-за чрезвычайно малой удельной поверхности исследованного образца, что неизбежно приводило к большой погрешности при определении Q. Хили и Фюрстенау [308] приводят значение теплоты смачивания для α-кварца ~0,З5 Дж/м2. Для других оксидов различия в теплотах смачивания, определенных разными авторами, также значительны. Для α-Al2O3 в [308] приведены значения Q 0,65 и 0,90 Дж/м2. Теплоты смачивания SnO2, приведенные в той же работе, составляют 0,40, 0,49 и 0,57 Дж/м2. Такой значительный разброс результатов имеет место и для других оксидов. Расхождения имеющихся в литературе данных по теплотам смачивания, безусловно, связаны с различным состоянием и предварительной подготовкой поверхности оксидов перед измерением. В работе [310] обсуждается зависимость теплот смачивания от температуры предварительной обработки твердого тела (Т). Зависимость Q – Т для кремнезема и оксида титана имеет следующий вид. С увеличением температуры прогрева образца величина Q сначала увеличивается, проходит через максимум, а затем уменьшается. Теплота смачивания оксида алюминия и оксида тория с увеличением температуры до 600°C растет [273,310]. Сложность зависимостей Q – Т для различных оксидов связана со сложностью и обратимостью процессов дегидратации и регидратации на поверхности оксида, что обсуждалось ранее при описании поверхностей оксидов (раздел II).
Размер частиц также оказывает влияние на величину Q. Как указывается в [310], теплота смачивания одного и того же оксида с ростом удельной поверхности уменьшается. Такое уменьшение было найдено для TiO2, SiO2, Al2O3.
Было замечено, что с ростом удельной поверхности образца SiO2 уменьшается его кристалличность. Авторы [310] считают, что разница в значениях Q связана с изменениями силового поля поверхности SiO2, которые зависят от его кристалличности. Таким образом, на теплоту смачивания оксидов оказывает влияние не только способ предварительной подготовки поверхности, но и размер частиц, и степень аморфизации поверхности.
В работе [308] обсуждалась связь величины теплоты смачивания оксидов с величиной точки нулевого заряда поверхности. По мнению авторов [308], теплота (энтальпия) смачивания и величина ТНЗ являются двумя основными характеристиками границы раздела оксид – вода, поскольку величина теплоты погружения (смачивания) дает прямой путь к термодинамике границы твердое тело – жидкость, а данные по ТНЗ требуются для решения основного уравнения электрохимии поверхности. Целью работы было показать, что основа соотношения между ТНЗ и термодинамическими свойствами поверхности оксид – жидкость, получаемыми из калориметрии теплот погружения, состоит в разностях сил электростатического поля твердой поверхности, которое в свою очередь может быть соотнесено с параметрами кристаллической решетки оксидов.
Стандартное уменьшение свободной энергии 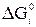 , сопровождающее процесс погружения (смачивания) сухого эвакуированного твердого неорганического оксида, составляет
, сопровождающее процесс погружения (смачивания) сухого эвакуированного твердого неорганического оксида, составляет
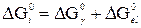 , (IV-1)
, (IV-1)
где 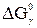 – вклад общей свободной энергии погружения, обусловленный изменением границы (граница с вакуумом заменяется границей с жидкостью), а
– вклад общей свободной энергии погружения, обусловленный изменением границы (граница с вакуумом заменяется границей с жидкостью), а 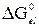 – вклад, обусловленный формированием ДЭС на поверхности.
– вклад, обусловленный формированием ДЭС на поверхности.
Ограничивая процесс образования заряда на поверхности реакцией
МеО– + 2Н+ ↔ МеОН2+ (IV-2)
c константой равновесия K, авторы [308] получают уравнение
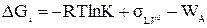 , (IV-3)
, (IV-3)
где 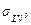 – свободная энергия границы между жидкостью и её насыщенным паром, а WA – работа адгезии жидкости к твердому телу.
– свободная энергия границы между жидкостью и её насыщенным паром, а WA – работа адгезии жидкости к твердому телу.
Соответственно,
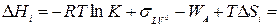
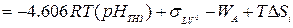 (IV -4)
(IV -4)
Считая работу адгезии постоянной практически для многих оксидов, а величину ΔSi меняющейся довольно незначительно, авторы [308] приходят к выводу, что
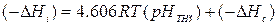 (IV-5)
(IV-5)
где Δ Н с – постоянная для серий оксид-вода, равная теплоте погружения оксида, ТНЗ которого находится при рН = 0.
Авторами было проведено сопоставление довольно большого количества литературных данных по Δ Нi и ТНЗ для оксидов с приблизительно одной и той же удельной поверхностью (~15 м2/г). Предварительная подготовка оксидов для калориметрических измерений проводилась при температуре 200°C и давлениях порядка 10-5мм Hg. Результаты сопоставления показали, что для кристаллических оксидов за некоторым исключением экспериментальные данные довольно хорошо ложатся на прямую линию в координатах Δ Нi – ТНЗ с наклоном 4,606 RТ для воды при 25°C, что подтверждает теоретические положения авторов [308].
Поскольку в настоящей работе исследуются процессы взаимодействия поверхности твердого тела с водными растворами электролитов, то, прежде чем перейти к рассмотрению экспериментальных данных по калориметрии смачивания водными растворами электролитов, необходимо остановится на некоторых вопросах термодинамики водных растворов.
IV.2. Структура водных растворов электролитов
Как известно, при растворении электролитов в воде происходят изменения структуры воды как в непосредственной близости к ионам, так и в объеме [311-313]. Такие изменения обусловлены взаимодействием ионов с молекулами воды, которые под действием поля иона так или иначе изменяют свое состояние и влияют на взаимодействие молекул воды между собой. Одним из первых, кто понял, что растворы нельзя рассматривать только как механические смеси растворителя с растворенным веществом, но как химические соединения, был Д.И.Менделеев [314].
В соответствии с воззрениями Самойлова [311], влияние ионов на структуру воды обусловлено, прежде всего, различием характера взаимодействия ионов с молекулами воды с одной стороны, и молекул воды между собой – с другой. Искажение ионами структуры воды при образовании раствора связано, во-первых, с появлением в воде частиц с радиусом, отличным от радиуса молекул воды; во-вторых, с различием координационного числа иона и молекулы воды; в третьих, с переориентацией ближайших к иону молекул воды и, в четвертых, с существованием двух состояний для молекул воды и наличием термодинамического равновесия между ними.
В растворе частицы перемещаются из одного положения равновесия в соседнее. При этом величина потенциальной энергии взаимодействующих частиц изменяется в зависимости от типа взаимодействия (диполь-дипольное, ион-дипольное и др.) Как показано в [311-313], поведение частицы в жидких растворах определяется тем вкладом, который вносится данным взаимодействием в величину потенциального барьера, преодолеваемого частицей. При рассмотрении свойств растворов в [315] учитывались силы, действующие на близких расстояниях (короткодействующие), между частицами, создающими структуру раствора.
Бернал и Фаулер [316], предложив тетраэдрическую модель жидкой воды, обратили внимание на те структурные, энергетические и химические изменения, которые наблюдаются в воде при добавлении к ней электролита. Маленькие ионы, обладающие большой плотностью заряда, и большие ионы с маленькой плотностью заряда по-разному воздействуют на структуру воды. Ионы в растворе создают собственную структуру и создаваемая структура проявляется в частности в том, что растворы по сравнению с чистой водой обладают большей или меньшей вязкостью. Структура, создаваемая слабогидратированными ионами (большими, с малым зарядом), дает вязкость раствора ниже вязкости чистой воды, более плотная структура, образованная сильногидратированными ионами, дает более высокую вязкость, чем в чистой воде. Бернал и Фаулер ввели понятие "структурная температура". Структурная температура ионного раствора определяется как температура, при которой структура и свойства чистой воды (вязкость, плотность) соответствуют структуре и свойствам ионного раствора при стандартной температуре. Ионы, имеющие небольшую плотность заряда, повышают структурную температуру (K+, Rb+, Cs+, Cl–, Br–, I– и др.), а ионы, обладающие более высокой плотностью заряда, понижают её (Li+, Na+, F–, SO42–, Mg2+, Al3+ и др.).
Кокс и Вольфенден [317] при исследовании вязкости растворов электролитов обнаружили, что растворы, содержащие ионы K+, Rb+, Cs+,NO3–, ClO3–, имеют вязкость ниже, чем в чистой воде.
Результаты по измерению вязкости полученные в [318-323] также однозначно показали, что ионы K+, Rb+, Cs+, ClO3–, NO3– характеризуются отрицательным коэффициентом вязкости и, следовательно, разрушают структуру воды. С увеличением температуры отрицательное значение коэффициента вязкости уменьшалось, а в отдельных случаях знак менялся на положительный.
Для ионов Na+, Li+, Mg2+, La3+ наблюдалось увеличение коэффициента вязкости, т.е. появление этих ионов в растворе способствовало укреплению структуры воды.
В [323] было обнаружено, что влияние электролитов на вязкость зависит от концентрации. Для 1н. растворов KCl и RbCl при 25°C наблюдалась отрицательная вязкость, а с увеличением концентрации вязкость увеличивалась и становилась больше, чем в чистой воде. Вязкость LiCl, NaCl при всех концентрациях была выше, чем в чистой воде. При больших разбавлениях вязкость растворов не отличается от вязкости чистой воды [319-320].
Для объяснения накопленных экспериментальных фактов по изменению вязкости, теплоемкости и других свойств растворов необходимо было разработать модель раствора электролита. Появились представления о гидратной оболочке, как оболочке, состоящей из двух слоев. За координационной сферой воды следует промежуточная сфера менее прочно связанных молекул воды, а на более далеких расстояниях от иона находятся молекулы свободного растворителя [324]. Согласно Френку и Эвансу [325], вокруг частиц в растворе существует три концентрические зоны. В первой зоне ионы ориентируют диполи воды с созданием вокруг иона довольно прочной оболочки. Вторая зона – промежуточная, где структура воды нарушена.
Во внешней зоне структура воды полностью сохраняется. Нарушение структуры воды во второй зоне зависит от природы иона.
Бокрис [326] общую сольватацию (взаимодействие иона с молекулами растворителя) разделял на два типа сольватации (гидратации). Первичная гидратация определялась, как прочное связывание молекул воды ионом. Вторичная гидратация определялась лишь электростатическим взаимодействием иона с молекулами воды, что приводит только к ориентации диполей воды. В современных работах [312,313] вместо терминов "первичная" и "вторичная" гидратация употребляются термины "ближняя" и "дальняя" гидратация.
По мнению Мищенко [327,328], для правильного понимания гидратации нужно учитывать всю сумму изменений, вызываемых появлением ионов в растворе.
Рассмотрение Самойловым гидратации ионов в водных растворах [311,312] основано на действии ионов на трансляционное движение ближайших к нему молекул воды раствора. При таком подходе рассматривается только "ближняя" гидратация. Что касается "дальней" гидратации, то она состоит, главным образом, в поляризации под действием поля иона окружающих молекул воды.
Особое значение при изучении "ближней" гидратации имеют так называемые кинетические свойства раствора, а именно: диффузия и самодиффузия, вязкость, теплопроводность, электропроводность, и др. Кинетические свойства растворов обусловлены малыми, но направленными возмущениями скачкообразного броуновского движения частиц (активированных скачков частиц от одного положения равновесия к соседнему). Активированные скачки частиц жидкости, как известно [211], определяются величинами потенциальных барьеров, разделяющих соседние положения равновесия, т.е. зависят от взаимодействия ближайших частиц жидкости.
Самойлов [311] предложил новый подход к изучению гидратации ионов. Он ввел величины ΔE i и τ i /τ, где ΔE i – величина изменения потенциального барьера, разделяющего соседние положения равновесия молекул воды, τ i – время пребывания молекулы воды в ближайшем к иону положению равновесия, а τ – время пребывания молекул воды в положении равновесия среди молекул воды. Величины ΔE i и τ i /τ являются достаточно общими количественными характеристиками гидратации. Если ион прочно связывает ближайшие молекулы воды, то τ i /τ велико. Уменьшение τ i /τ означает ослабление связи иона с ближайшими молекулами воды раствора. В соответствии с влиянием ионов на вязкость раствора они так же действуют на трансляционное движение ближайших к ним молекул воды. Ионы Mg2+, Ca2+, Li+ ослабляют это движение, ионы K+, Cs+, I– усиливают трансляционное движение. Таким образом, существует два случая при взаимодействии иона с молекулами воды:
1) ΔE i > 0 и τ i /τ > 1;
2) ΔE i < 0 и τ i /τ < 1.
Первый случай соответствует эффективному связыванию ионами ближайших молекул воды. Во втором случае молекулы воды вблизи ионов становятся более подвижными, чем в чистой воде. Это явление было названо Самойловым "отрицательной" гидратацией. Рассуждения Самойлова находятся в полном соответствии с представлениями Бернала и Фаулера [316] о структурной температуре. Ионам, повышающим структурную температуру, как раз и свойственна отрицательная гидратация.
Для описания структуры концентрированных растворов электролитов Мищенко и Сухотин [329] ввели понятие "граница полной сольватации". С увеличением концентрации при достижении границы полной сольватации в растворе исчезают зоны собственной структуры воды и деструктурированной воды. Граница полной сольватации достигается при такой концентрации электролита, при которой все молекулы растворителя распределяются между сольватными оболочками катионов и анионов. Ионы в растворе ведут конкурентную борьбу за растворитель, молекулы которого подвергаются перераспределению в зависимости от гидратационной способности ионов.
Участок от бесконечного разбавления до границы полной сольватации рассматривается в [330] как растворитель, искаженный присутствием ионов. Второй участок – от этой границы сольватации до насыщения – как электролит, искаженный присутствием растворителя. Самойлов [311,331] выделяет три зоны, рассматривая весь интервал концентраций от разбавленного до насыщенного. Для насыщенного раствора, находящегося в равновесии с твердой солью, сохраняется строение, соответствующее кристаллогидрату, а ионы имеют координационное число, свойственное кристаллической решетке. По мере разбавления раствора доминирующая роль влияния электролита падает, и всё сильнее проявляется определяющее влияние квазикристаллической структуры воды, достигающее наибольшего значения для разбавленных систем.
Сато [332] использовал модель Фрэнка и Вина [333] для рассмотрения концентрированных растворов. По модели [332] по мере увеличения концентрации электролита расстояния между гидратированными ионами уменьшаются за счет уменьшения объема, занимаемого структурно-нормальной водой. При концентрациях 1,5-2,0 М гидратные оболочки перекрываются. Область структурно нормальной воды перестает существовать (достигается, по определению Мищенко [329], граница полной сольватации). При концентрациях, больше, чем 2М, строение раствора напоминает расплав солей, сохраняющих свою кристаллическую решетку.
Основными величинами, дающими возможность установить гидратацию иона в растворе, являются числа гидратации и координационные числа. При определении чисел гидратации различными методами наблюдаются расхождения на один или два порядка [334,335]. Существующие методы определения чисел гидратации пока не в состоянии однозначно решить вопрос, касающийся нахождения энергетической границы для связанных и не связанных ионом молекул воды. Анализ методов определения чисел гидратации и оценка полученных величин даются в работах [336-338]. Определение координационных чисел было проведено в работах [327,328,339-341]. Анализ указанных работ показывает, что координационные числа могут быть как целыми, так и дробными. Дробные величины отвечают некоторому усредненному структурному порядку.
Координационные числа ионов щелочных металлов близки к четырем [342,343], для двухзарядных ионов в разбавленных растворах координационные числа приближаются к шести [327,328,339,340]. Имеются попытки теоретических расчетов координационных чисел и энергии взаимодействия между различными структурными единицами раствора, например, с помощью метода Монте-Карло [344]. С повышением концентрации координационное число ионов в растворе изменяется под действием полей других ионов, присутствующих в растворе. В таких растворах могут появиться структуры кристаллогидратов [345].
Поскольку взаимодействие между ионом и молекулами растворителя оказывает влияние на энергетические характеристики системы, большое внимание в изучении растворов электролитов всегда уделялось калориметрическим исследованием, в частности, теплот растворения солей и других соединений. Из калориметрических данных рассчитывались теплоты гидратации и оценивались значения энтропии гидратации. В [346] приводятся значения энтропии и теплоты гидратации для 34 катионов. Позднее в работах Крестова с сотрудниками многие значения были уточнены [347-349]. Было обнаружено, что величина энтропии для ряда щелочных катионов от Li+ до Cs+ меняет знак с отрицательного на положительный [312,313]. Граница изменения знака проходит между ионами Na+ и K+. Увеличение изменения энтропии свидетельствует о преобладании при растворении разупорядочивающего эффекта.
В многочисленных работах Крестова с сотрудниками изучается изменение термодинамических характеристик в процессе гидратации ионов от различных факторов. Изучались структурные составляющие энтропийных характеристик растворения галогенидов щелочных и щелочноземельных металлов в обычной и тяжелой воде [350,351], термодинамика гидратации ионов при различных температурах [352,353,354], детально рассматривались энтропийные характеристики ближней и дальней гидратации [348]. Было обнаружено, что в области дальней гидратации изменение энтропии меньше нуля и, следовательно, дальняя гидратация есть положительная гидратация. Изменение энтропии ближней гидратации может быть как положительным, так и отрицательным. В [349] Крестовым была введена величина ΔSII (изменение энтропии, связанное со структурными изменениями воды при гидратации ионов). Изменение величины ΔSII связано с двумя типами изменений структуры воды. Во-первых, действие поля иона приводит к нарушению упорядоченности молекул воды, характерной для чистой воды. Этот эффект сопровождается увеличением энтропии. Во-вторых, действие поля иона приводит к ориентации молекул воды в этом поле, что ведет к упорядочиванию и, следовательно, к уменьшению энтропии. Общий знак изменения энтропии, связанного со структурными изменениями воды под действием поля иона, зависит от преобладающего влияния одного из этих эффектов. Анализ изменения ΔSII в зависимости от радиуса иона показывает, что перемена знака ΔSII с положительного на отрицательный для однозарядных катионов наблюдается при r ~ 1,1Å, для двухзарядных – при r ~ 1,7Å, для трехзарядных – при r ~ 1,9Å. Для анионов в [349] также была получена соответствующая зависимость изменения величины ΔSII.
Вопросы взаимовлияния различных ионов на гидратацию исследовались в серии работ Самойлова и его сотрудников [355-357] по теории высаливания из водных растворов. Было показано, что основное значение имеет ориентация молекул воды вблизи высаливаемого иона и её изменение под действием ионов высаливателя. Катионы высаливателя уменьшают, анионы – увеличивают ближнюю гидратацию высаливаемого катиона.
При исследовании двойных и тройных систем вода – этанол (этиленгликоль), вода – этанол – этиленгликоль Егоровой [358] были получены интересные данные по теплотам растворения (Δ H) KCl. Было обнаружено, что этиленгликоль понижает Δ H, а этиловый спирт повышает. Такой эффект связан с тем, что этиловый спирт до определенной концентрации стабилизирует структуру воды, а этиленгликоль разрушает её во всей области концентраций.
Вопросы термодинамики растворения для растворов различных концентраций также были подробно изучены [353,354,359]. Было, в частности, обнаружено довольно сложное поведение теплоты растворения некоторых галогенидов щелочных металлов при очень низких концентрациях (наличие максимумов и минимумов на зависимости теплоты растворения от концентрации) [353,354].
Несмотря на очень большое число работ по вопросам гидратации ионов, вопросы количественной характеристики положительной и отрицательной гидратации нельзя считать окончательно решенными.
IV.3. Экспериментальные исследования иммерсионного смачивания
IV.3.1. Измерения зависимости теплот смачивания на кварце от
концентрации и состава раствора неорганических электролитов.
В качестве объекта исследования для изучения влияния состава раствора электролитов на теплоту смачивания оксидов был выбран один из наиболее изученных во всех отношениях оксидов – оксид кремния. Исследовались две модификации кремнезема: кристаллический α-кварц Кыштымского месторождения и аморфная модификация SiO2 (так называемый плавленый кварц марки КСГ, Ленинград). Исходные образцы, имеющие крупность зерен 0,3-1,0 мм, подвергались очистке кислотой (HCl) для удаления соединений железа, попавших при помоле в стальных мельницах, отмыванию водой и высушиванию. Известно [16,47], что степень аморфизации поверхности твердого тела при помоле существенно зависит не только от способа измельчения (вибропомол, истирание, ударный помол), но и от среды, в которой производится измельчение. Для того, чтобы свести к минимуму возможные изменения структуры поверхности при помоле, а также в результате последующих операций очистки (обработка кислотой и отмывка водой), исходные образцы были подвергнуты сухому ударному помолу в мельнице с покрытием из карбида вольфрама марки ТЕМА (ФРГ), практически не дающему намола. После помола кварц дальнейшей очистке не подвергался и не был в контакте с водой до калориметрических измерений. Химический состав исследованных образцов кварца после помола по результатам анализа, выполненного по стандартным методикам, применяемым для анализа кремнезема [360] (таблица в приложении), показал высокую чистоту полученных образцов (содержание SiO2 99,38% для α-кварца и 99,55% для КСГ) и пренебрежимо малое содержание примесей.
Рентгеноструктурный анализ образцов кварца, выполненный с помощью дифрактометра ДРОН-3 (излучение кобальтовое, отфильтрованное от β-серии железной пластинкой-фильтром) позволил сделать вывод, что в процессе помола кристаллическая структура α-кварца не была нарушена, а плавленый кварц КСГ действительно является аморфной модификацией кремнезема. Было также проведено исследование молотых образцов кварца методом электронного парамагнитного резонанса, показавшее отсутствие на поверхности кварца заметных количеств свободных радикалов [32].
Удельная поверхность полученных образцов кварца была определена методом БЭТ по низкотемпературной адсорбции азота. Она составляла 3,9 м2/г для кристаллического кварца и 5,2 м2/г для плавленого.
Калориметрические измерения проводили на отечественном микрокалориметре ДАК-1-1А по методике, приведенной в приложении. Для проведения измерений в узкой ячейке калориметра ДАК-1-1А нами была разработана новая конструкция ампулы [361], позволявшая с достаточной точностью стандартизировать так называемую теплоту разбивания ампулы [15].
Уже первые опыты по изучению зависимости теплот смачивания от состава раствора на кристаллическом кварце показали, что теплота смачивания зависит как от концентрации потенциал определяющих ионов (рН раствора) так и от содержания индифферентного электролита. В соответствии с этим в дальнейшем было проведено две серии измерений. В первой серии исследовалось влияние рН раствора на теплоту смачивания (Q) для чистых растворов HCl, KOH или LiOH, а также в присутствии постоянного фона (10–2 н.) хлоридов щелочных и щелочноземельных металлов. Полученные результаты приведены на рис. 46а и б.
|
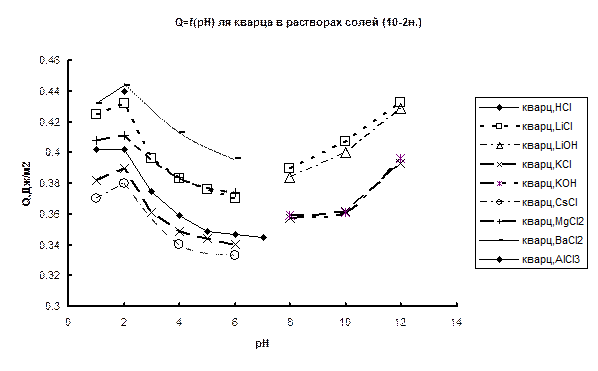
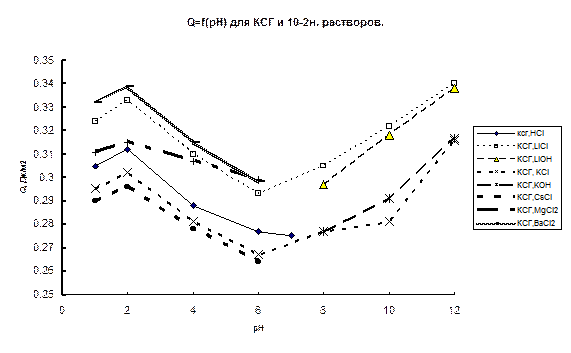
Рис.46. Зависимости теплоты смачивания кварца (а) и аморфного кремнезема марки КСГ (б) от pH раствора без солевого фона и в его присутствии (10-2н.).
В другой серии измерений исследовалась зависимость теплоты смачивания от концентрации нейтральных растворов изученных хлоридов. Результаты этой серии измерений приведены на рис.47а и б.
|
|
|
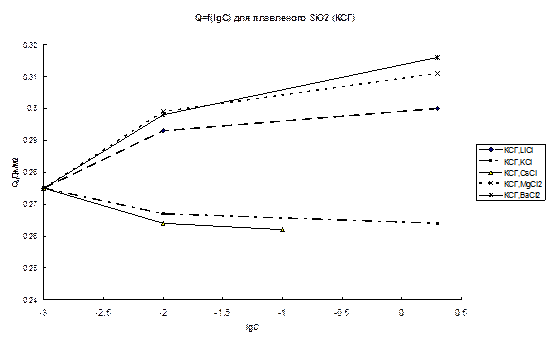
Рис.47. Зависимости теплоты смачивания кристаллического (кварц) (а) и аморфного (КСГ) (б) SiO2 от концентрации раствора.
В статье Гриффитса и Фюрстенау [273], вышедший из печати после завершения первой части нашего исследования, было проведено изучение зависимости теплоты смачивания α-Al2O3 от рН раствора (HCl, NaOH, солевой фон 2·10-3 н. NaCl) Общий вид полученной зависимости в общем аналогичен полученным нами результатам, c той разницей, что минимум теплоты смачивания находился при рН, соответствующем величине ТНЗ α-Al2O3 (рН 8,8).
Полученное изменение теплового эффекта авторы [273] трактовали как влияние энтальпии образования ДЭС на теплоту смачивания.
Возвращаясь к полученным нами данным, отметим, что характер смещения кривых Q – рН в присутствии солевого фона относительно кривой Q – рН для НС1 (рис. 46) качественно согласуется с влиянием катиона соли на структуру воды [311-313]. Усиливающие структуру воды Li+, Mg2+, Ba2+, Al3+ увеличивают теплоты смачивания, разрушающие структуру воды K+ и Cs+ – уменьшают. Аналогичное влияние оказывает солевой фон и в щелочной области для K+ и Li+. Величина изменения теплоты смачивания при росте рН от 2 до 6 (Δ Q d = Q pH2 – Q pH6) очень мало изменяется для HCl, KCl и LiCl. Остальные катионы значительно больше влияют на величину Δ Q d, что связано, по-видимому, с их известной из электрокинетических измерений специфичностью по отношению к поверхности кварца [6,7]. Обращает на себя внимание, что величина изменения теплоты смачивания при добавлении соли (Δ Q S) для щелочных металлов довольно слабо зависит от рН раствора, однако наблюдается небольшая тенденция к уменьшению абсолютной величины этого эффекта при переходе от рН 2 к рН 6. Интересно отметить, что соотношение величин средних смещений (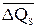 ) кривых Q – рН для KCl и LiCl относительно кривой для чистой HCl (рис.46, сравнение ординат при данном рН кривых 2 и 4 с ординатами кривой 1) находится в количественном согласии с соотношением теплот растворения (Δ H S) KCl и LiCl для концентраций 10–2 н. (
) кривых Q – рН для KCl и LiCl относительно кривой для чистой HCl (рис.46, сравнение ординат при данном рН кривых 2 и 4 с ординатами кривой 1) находится в количественном согласии с соотношением теплот растворения (Δ H S) KCl и LiCl для концентраций 10–2 н. (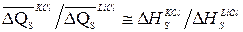 ). Сравнение
). Сравнение 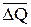 для растворов MgCl2 и BaCl2 с теплотами растворения этих солей в воде показывает, что они коррелируют по знаку, но не по величине, что может быть связано со специфичностью взаимодействия катионов этих солей с поверхностью кварца. Обнаруженные факты свидетельствуют о том, что все исследованные соли оказывают влияние на состояние граничных слоев воды на поверхности кварца.
для растворов MgCl2 и BaCl2 с теплотами растворения этих солей в воде показывает, что они коррелируют по знаку, но не по величине, что может быть связано со специфичностью взаимодействия катионов этих солей с поверхностью кварца. Обнаруженные факты свидетельствуют о том, что все исследованные соли оказывают влияние на состояние граничных слоев воды на поверхности кварца.
Как видно из рис. 47, увеличение концентрации солей приводит к росту или падению теплот смачивания также в соответствии с влиянием катиона на структуру воды. При этом зависимости Q – lgC для хлоридов щелочных металлов в случае кристаллического кварца практически линейны. Для проверки, оказывают ли на теплоту смачивания влияние только катионы или вся соль в целом, были проведены опыты с LiClO4. Как известно, ClO4–, в отличие от Li+, оказывает значительное разрушающее действие на структуру воды. Из рис.47 видно, что для LiClO4, в отличие от LiCl, наблюдается довольно слабая зависимость теплоты смачивания от концентрации, что легко объясняется взаимно компенсирующим влиянием аниона и катиона на структуру воды. Полученная зависимость подтверждает предположение о влиянии на граничный слой как катиона, так и аниона соли. Очевидно, что обмен ионов в ДЭС (замена Н+-иона силанольных групп поверхности на катион соли) также может оказывать влияние на суммарный тепловой эффект. Однако тот факт, что влияние соли довольно слабо зависит от рН раствора в области рН 1-6, т.е. от степени диссоциации силанольных групп, в сочетании с обнаруженным влиянием аниона на зависимость Q – lgC позволяет предположить, что обмен ионов в ДЭС для данной области рН не является определяющим фактором зависимости Q – lgC.
В соответствии с условиями эксперимента, в разных опытах при контакте поверхности твердого тела с раствором граничный слой воды (нерастворяющий объем) формируется из раствора различной концентрации. Известно [10-13], что величина нерастворяющего объема, определяемая по изменению концентрации раствора после взаимодействия с поверхностью твердого тела, зависит от концентрации раствора. Очевидно, что при формировании нерастворяющего объема происходит перераспределение растворенного электролита между объемом раствора и граничным слоем. При этом происходит также формирование ДЭС, сопровождающееся обменом ионов раствора с исходной поверхностью кварца. Как степень вхождения электролита в граничный слой, так и обмен ионов зависят от концентрации раствора. Известно также [328,358], что теплоты растворения солей зависят от состава растворяющей среды. Авторы [358] обнаружили, что добавление в воду этилового спирта, укрепляющего структуру воды, увеличивает эндотермический эффект растворения KCl, а добавление оказывающего разрушающее действие этиленгликоля понижает. По-видимому, обнаруженная нами зависимость теплот смачивания от типа и концентрации растворенного электролита представляет собой явление того же порядка. Зависящее от концентрации раствора вхождение электролита в граничный слой представляет собой изменение среды распределения электролита и приводит к изменению энтальпии системы, пропорциональному lgC.
На рис. 46б и 47б приведены зависимости Q – рН и Q – lgC для плавленного кварца. Как видно из рисунков, все обнаруженные для кристаллического кварца зависимости теплот смачивания сохраняются, но абсолютные величины всех эффектов существенно ниже. Кроме того, для плавленого кварца зависимости Q – рН имеют гораздо более пологий наклон в области рН 2–4. Гораздо меньшие средние величины теплот смачивания для аморфного SiO2 говорят о более слабом влиянии его поверхности на структуру воды. В таблице 8 приведены величины отношения теплот смачивания кристаллического кварца Q c к теплотам смачивания аморфного SiO2 (Q a) для разных величин рН и состава раствора.
Таблица 8
Величина Q c/ Q a для разных рН и состава раствора.
| Раствор | 10–2 н. рН 2 | 10–2 н. рН 1 | 10–2 н. рН 4 | 10–2 н. рН 6 | 10–3 н. рН 6 | 10–1 н. рН 6 | 2 н. рН 6 |
| HCl* | 1,29 | 1,32 | 1,25 | 1,25 | – | – | – |
| LiCl | 1,29 | 1,31 | 1,24 | 1,24 | – | – | 1,33 |
| KCl | 1,29 | 1,29 | 1,24 | 1,27 | – | – | 1,26 |
| CsCl | 1,29 | 1,28 | 1,22 | 1,26 | – | 1,24 | – |
| MgCl2 | 1,30 | 1,31 | 1,25 | 1,25 | – | – | 1,25 |
| BaCl2 | 1,31 | 1,30 | 1,31 | 1,33 | – | – | 1,31 |
| LiClO4 | – | – | – | – | 1,31 | 1,28 | – |
* без солевого фона
Как видно из таблицы, на отношение Q c/ Q a состав раствора почти не оказывает влияния. Наименьшее влияние на величину Q c/ Q a тип электролита оказывает, как и следовало ожидать, в ТНЗ кварца. Развитие ДЭС (рН > 2) приводит к некоторому уменьшению этой величины, однако в целом изменение отношения Q c/ Q a невелико (1,25-1,30). Это свидетельствует о том, что определяющим при образовании граничных слоев фактором является структура твердого тела. Интересно отметить, что величина Q c/ Q a практически совпадает с величиной отношения констант Гамакера (равного 1,3) для кристаллической и аморфной модификации SiO2,. Изменение рН и состава раствора вызывает изменения, пропорциональные главному фактору, следствием чего и является практическое постоянство Q c/ Q a.
Таблица 9
Величины 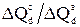 для разных участков изменения рН
для разных участков изменения рН
| Раствор | рН 2-6 | рН 2-4 | рН 4-6 |
| HCl | 1,54 | 1,79 | 1,09 |
| LiCl | 1,55 | 2,13 | 0,76 |
| KCl | 1,42 | 2,16 | 0,56 |
| CsCl | 1,52 | 2,35 | 0,50 |
| MgCl2 | 2,31 | 3,38 | 1,25 |
| BaCl2 | 1,17 | 1,25 | 1,00 |
В таблице 9 приведены величины отношения для разных участков изменения рН. Данные этой таблицы показывают, что процесс диссоциации силанольных групп на кристаллическом и аморфном SiO2 проходит, по-видимому, по-разному. Особенно сильное различие наблюдается в области рН 2-4, однако и для всего интервала pH в кислой области (2-6) диссоциация силанольных групп гораздо интенсивнее проходит на кристаллическом SiO2 (отношение значительно больше отношений Q c/ Q a), т.е. на процесс диссоциации силанольных групп структура кварца оказывает наиболее сильное воздействие. Это согласуется с известным фактом бóльших величин ζ-потенциала для кристаллического кварца по сравнению с аморфным при прочих равных условиях [363]. Обнаруженный факт более интенсивной диссоциации силанольных групп на поверхности кристаллического кварца, особенно в области рН 2-4 позволяет предположить, что константа диссоциации этих групп для кристаллического кварца отличается от константы диссоциации силанольных групп аморфного кремнезема, причем первая находится в более кислой области, чем вторая. Во всяком случае величина рН, соответствующая 1/2Δ Q d для кристаллического карца составляет 3,2, а для аморфного 3,6.
Как уже упоминалось, при взаимодействии твердого тела с раствором на поверхности раздела фаз образуются граничные слои воды с измененными свойствами. При этом на поверхности образуется двойной электрический слой. При взаимодействии поверхности твердого тела с раствором в граничном слое происходит перераспределение концентрации растворенного вещества. Таким образом, теплота смачивания является интегральной величиной, относящейся к слою жидкости у поверхности твердого тела и включающей в себя тепловые эффекты целого ряда процессов. При этом, по способу определения, теплота смачивания является избыточным эффектом, основная часть которого характеризует изменение энтальпии граничного слоя воды в реакциях взаимодействия с поверхностью твердого тела. При образовании ДЭС на поверхности SiO2 могут идти следующие реакции:
1. –SiOH ↔ –SiO– + H+ (+Δ H)
2. –SiOH + H+ ↔ –SiOH2+ (-Δ H)
3. H+ + OH– ↔ H2O (-Δ H)
4. –SiOH + Me+ ↔ –SiOMe + H+ (±Δ H)
Реакции диссоциации (1) идут с поглощением тепла, реакции присоединения (2,3) — с выделением.
Поскольку при взаимодействии раствора с поверхностью твердого тела может происходить перераспределение растворенного вещества между граничным слоем и объемом раствора, а  также растворение твердого тела, для уточнения протекающих процессов необходимо было изучить изменение состава раствора после взаимодействия его с поверхностью твердого тела.
также растворение твердого тела, для уточнения протекающих процессов необходимо было изучить изменение состава раствора после взаимодействия его с поверхностью твердого тела.
IV.3.2. Изменение концентрации смачивающего раствора и
граничные слои жидкости на кварце
Было проведено изучение изменения рН раствора, содержания ионов Li+, K+, Cl– в растворах LiCl и KCl, а также количества растворенного кремнезема после взаимодействия раствора с порошками кристаллического и плавленого кварца.
Определение концентрации ионов Li+ и K+ проводилось методом фотометрии пламени на приборе FLAPHO (ГДР), содержание растворенного кремнезема определялось фотокалориметрическим методом, разработанным на кафедре аналитической химии ЛГУ. Cl–-ион определялся меркуриметрическим титрованием. Проводилось также потенциометричес-кое титрование порошков кварца растворами НС1 и KOH.
Соотношение массы твердой и жидкой фаз в этих опытах составляло 1:30 и соответствовало опытам по теплотам смачивания. Для растворов
 10-2н. KCl и LiCl при таком соотношении фаз аналитические опыты не показали измеримого изменения концентрации Li+, K+ и Cl–. Для выяснения характера взаимодействия растворов LiCl и KCl с поверхностью кварца были проведены также опыты с растворами 10-3н. при соотношении массы твердой и жидкой фаз, равном 1:3 (5г. кварца и 15 мл раствора). Опыты проводились после контакта раствора с порошком в течение 1,5 часов (время калориметрических измерений при изучении теплот смачивания). Через 1,5 часа твердая фаза отделялась центрифугированием, а полученный раствор анализировался.
10-2н. KCl и LiCl при таком соотношении фаз аналитические опыты не показали измеримого изменения концентрации Li+, K+ и Cl–. Для выяснения характера взаимодействия растворов LiCl и KCl с поверхностью кварца были проведены также опыты с растворами 10-3н. при соотношении массы твердой и жидкой фаз, равном 1:3 (5г. кварца и 15 мл раствора). Опыты проводились после контакта раствора с порошком в течение 1,5 часов (время калориметрических измерений при изучении теплот смачивания). Через 1,5 часа твердая фаза отделялась центрифугированием, а полученный раствор анализировался.
Рис.48. Кривая потенциометрического титрования кварца(10-2н. KCl, титрование HCl и KOH)ю1 – холостое титрование, 2 – титрование образца кварца.
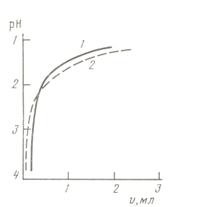
Изменение рН раствора после взаимодействия с кварцем контролировалось после опытов по определению теплот смачивания и с помощью потенциометрического титрования. Оба метода показали одинаковый характер изменения рН раствора после взаимодействия с кварцем. На рис. 48 представлена кривая потенциометрического титрования кристаллического кварца в растворе 10-2н. KCl (для плавленого кварца кривая потенциометрического титрования имеет аналогичный вид). Из рис. 48 видно, что в результате взаимодействия порошка кварца с раствором в области рН≤2 наблюдается подкисление раствора, а при рН>2 подщелачивание. Точка нулевого заряда (ТНЗ) находится при рН 2,08 для кристаллического и при рН 2,10 для плавленого. Обнаруженное изменение рН раствора в кислой области правее и левее ТНЗ противоречит, на первый взгляд, общепринятому механизму образования заряда на поверхности кварца. Согласно этому механизму, в области рН<2 на поверхности кварца происходит сорбция ионов Н+, поверхность заряжается положительно, а рН взаимодействующего раствора увеличивается (подщелачивание). При рН>2 на поверхности начинается диссоциация силанольных групп, поверхность заряжается отрицательно, а рН раствора уменьшается (подкисление). Прежде чем перейти к обсуждению нестандартного хода кривой потенциометрического титрования, рассмотрим, как изменяется концентрация ионов Li+ и K+ после взаимодействия растворов хлоридов калия и лития с порошком кварца.
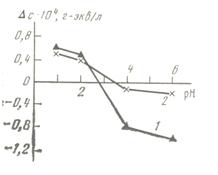 Рис.49. Изменение концентрации катиона после взаимодействия раствора с кварцем в зависимости от pH раствора. 1 – KCl, 2 – LiCl.
Рис.49. Изменение концентрации катиона после взаимодействия раствора с кварцем в зависимости от pH раствора. 1 – KCl, 2 – LiCl.
На рис. 49 представлены результаты по зависимости изменения концентрации (Δ c) калия и лития после взаимодействия исходного раствора с кристаллическим кварцем от рН раствора. Как видно из рис. 49, при рН≤2 наблюдается увеличение концентрации ионов Li+ и K+, а при рН>3,5 — уменьшение. Таким образом, сопоставление данных, представленных на рис. 48 и 49 показывает, что при рН≤2 наблюдается увеличение концентрации всех катионов в системе кварц — раствор электролита, а при рН от 3,5 до 6 — уменьшение.
Увеличение содержания катиона при рН≤2 после взаимодействия с кварцем можно трактовать как предпочтительную сорбцию воды (по сравнению с электролитом) на поверхности кварца — образование нерастворяющего объема [11]. Формирование нерастворяющего объема приводит к увеличению концентрации растворенного вещества в свободном объеме. Наблюдаемое при рН от 3,5 до 6,0 уменьшение концентрации Li+ и K+ в растворе в значительной степени объясняется, по-видимому, ионным обменом на поверхности кварца. Увеличение рН при этом связано, по всей видимости, с известным в литературе фактом связывания HCl различными формами растворенного кремнезема [364]. Как уже упоминалось [11]; величина нерастворяющего объема на данной поверхности зависит от вида и концентрации растворенного вещества. Можно предположить, в согласии с литературными данными [362], что при рН>2 происходит некоторое утоньшение граничного слоя, т.е. уменьшение нерастворяющего объема, что также оказывается на характере распределения растворенного вещества в системе.
 Из данных, представленных на рис. 49, видно, что изменение концентрации Li+ в растворе гораздо слабее, чем K+. Этот экспериментальный факт может быть объяснен тем, что структурирующее действие Li+ на воду сказывается в некотором увеличении граничного слоя воды в системе кварц — раствор LiCl по сравнению с KCl. К сожалению, данные аналитического определения изменения концентрации раствора в результате взаимодействия с поверхностью позволяют только качественно судить об изменении соотношения электролит — вода в граничном слое. Очевидно, однако, что при одном и том же количестве катиона металла на поверхности твердого тела, появившегося в результате обмена с H+-ионами силанольных групп, но при разной протяженности граничного слоя результат аналитического определения концентрации измененного раствора будет различен. Если пренебречь объемом граничного слоя, из соотношения массы и удельной поверхности кварца и объема исходного раствора и из величины изменения концентрации катиона после взаимодействия с кварцем можно оценить количество Li+ и K+, перешедших в граничный слой[1] (на единицу поверхности). Для K+ при рН 6 эта величина составляет ~4·1016 ионов на м2, а для Li+ 1·1016 м-2. Различие в 4 раза намного превышает погрешности определения и расчета и безусловно свидетельствует о различном влиянии K+ и Li+ на граничный слой.
Из данных, представленных на рис. 49, видно, что изменение концентрации Li+ в растворе гораздо слабее, чем K+. Этот экспериментальный факт может быть объяснен тем, что структурирующее действие Li+ на воду сказывается в некотором увеличении граничного слоя воды в системе кварц — раствор LiCl по сравнению с KCl. К сожалению, данные аналитического определения изменения концентрации раствора в результате взаимодействия с поверхностью позволяют только качественно судить об изменении соотношения электролит — вода в граничном слое. Очевидно, однако, что при одном и том же количестве катиона металла на поверхности твердого тела, появившегося в результате обмена с H+-ионами силанольных групп, но при разной протяженности граничного слоя результат аналитического определения концентрации измененного раствора будет различен. Если пренебречь объемом граничного слоя, из соотношения массы и удельной поверхности кварца и объема исходного раствора и из величины изменения концентрации катиона после взаимодействия с кварцем можно оценить количество Li+ и K+, перешедших в граничный слой[1] (на единицу поверхности). Для K+ при рН 6 эта величина составляет ~4·1016 ионов на м2, а для Li+ 1·1016 м-2. Различие в 4 раза намного превышает погрешности определения и расчета и безусловно свидетельствует о различном влиянии K+ и Li+ на граничный слой.
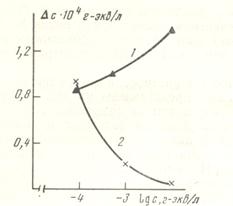
Рис. 50. Изменение концентрации катиона после взаимодействия кварца с растворами различных начальных концентраций: 1 – KCl, 2 – LiCl.
На рис. 50 приведены данные по изменению концентрации ионов Li+ и K+ после взаимодействия с кварцем в зависимости от начальной концентрации нейтральных растворов KCl и LiCl (pH~6). Как видно из рис. 50, сорбция K+ увеличивается с увеличением исходной концентрации раствора, сорбция Li+ уменьшается. Обнаруженное явление можно объяснить тем, что с увеличением концентрации Li+ возрастает его структурирующее действие в граничном слое воды. Толщина адсорбированных слоев воды на поверхности кварца растет по мере возрастания концентрации LiCl, что сказывается на концентрации свободного объема раствора. Увеличение развитости граничного слоя частично компенсирует поглощение Li+ из объема на поверхность вследствие ионного обмена. Увеличение концентрации КС1 приводит к увеличению деструктурирующего действия иона K+ в поверхностном слое воды, что способствует уменьшению граничного слоя. В результате этого суммарная сорбция электролита в поверхностном слое увеличивается. При концентрации KCl и LiCl 10-4н. изменения концентрации ионов Li+ и K+ практически одинаковы, что может быть связано с одинаковым развитием поверхностных слоев воды при такой низкой концентрации электролита.
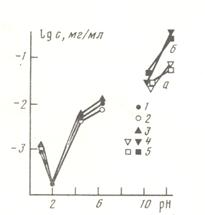 Рис. 51. Количество растворенного кремнезема в зависимости от pH раствора: 1 – HCl, кварц; 2 – HCl, аморфный SiO2 (КСГ); 3 – KCl, кварц; 4 – KOH; 5 – LiOH; а – кварц, б – КСГ.
Рис. 51. Количество растворенного кремнезема в зависимости от pH раствора: 1 – HCl, кварц; 2 – HCl, аморфный SiO2 (КСГ); 3 – KCl, кварц; 4 – KOH; 5 – LiOH; а – кварц, б – КСГ.
Результаты определения количества растворенного кремнезема в зависимости от рН и состава раствора представлены на рис. 51. Следует отметить, что полученные нами результаты не относятся к состоянию равновесия, т.к. скорость растворения кварца мала и равновесие достигается через несколько суток [365-368]. В нашем случае количество кремнезема, перешедшего в раствор за время контакта (1,5 часа) минимально при рН 2, т.е. в ТНЗ кварца. При pH 1, и при рН>2 концентрация кремнезема увеличивается. Такая зависимость от рН раствора объясняется тем, что при отклонении рН от ТНЗ, т.е. при развитии заряда на поверхности (образование ДЭС) усиливается взаимодействие поверхности с диполями воды и количество кремнезема в растворе увеличивается. Количество растворенного кристаллического кремнезема при рН 4 и 6 несколько выше, чем плавленого. В растворе KCl количество растворенного кремнезема несколько возрастает. Возможно, в данном случае играют роль кинетические факторы, когда катион обладает способностью ускорять процесс достижения равновесия [365]. В растворах LiOH и KOH количество растворенного кремнезема резко возрастает. Механизм растворения, описанный в [366], учитывает каталитическую роль иона ОН–, который способен хемосорбироваться на поверхности кремнезема и повышать координационное число поверхностных атомов кремния более четырех, ослабляя, таким образом, кислородные связи с лежащими ниже атомами кремния.
Поскольку растворение кварца, как известно [366,369], сопровождается значительными тепловыми эффектами, нами была сделана попытка учесть теплоту растворения кварца для того, чтобы выделить ту часть теплового эффекта, которая соответствует собственно теплоте смачивания.
Количество растворенного кварца в воде при 25°C составило 5,46·10-7 моль/м2 (в пересчете на SiO32- на м2 поверхности кварца. Энтальпия растворения кварца, согласно [370], составляет +25,1·103Дж/моль. Количество теплоты, соответствующее растворению вышеупомянутого количества кварца составляет 0,014 Дж/м2. Таким образом, истинное значение теплоты смачивания кристаллического кварца водой должно быть равно 0,347 Дж/м2 (измеренная теплота смачивания) + 0,014 Дж/м2 = 0,361 Дж/м2. Поскольку растворение кварца связано с разрушением его кристаллической решетки, можно предположить, что энтальпия растворения кварца в растворах электролитов близка к энтальпии растворения его в воде. Используя это допущение, можно оценить исправленное значение теплоты смачивания во всем интервале рН. Аналогичный расчет поправок на теплоту растворения был сделан для плавленого кварца. Значение энтальпии растворения плавленого кварца согласно [365,370] составляет +11,1 103Дж/моль.
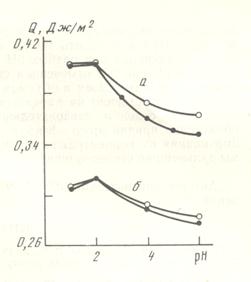 Рис.52. Зависимость теплоты смачивания от рН раствора HCl кварца (а) и аморфного SiO2 (КСГ) (б): 1 – измеренные теплоты смачяивания, 2 – теплоты смачивания с поправкой на растворение кварца.
Рис.52. Зависимость теплоты смачивания от рН раствора HCl кварца (а) и аморфного SiO2 (КСГ) (б): 1 – измеренные теплоты смачяивания, 2 – теплоты смачивания с поправкой на растворение кварца. В щелочной области, особенно при рН 12, величина поправки на растворение кварца резко увеличивается как для кристаллического, так и для плавленого кварца. При рН 12 величина поправки составляет уже 0,336 Дж/м2 для раствора KOH и 0,363 Дж/м2 для раствора LiOH. Если сравнить полученные величины поправки со значением теплот смачивания кристаллического кварца растворами KOH (0,395 Дж/м2) и LiOH (0,429 Дж/м2), то видно, что величина поправки достигает 70-80% от измеряемого в калориметре эффекта и с учетом этой поправки теплота смачивания в щелочных растворах должна достигать 0,7-0,8 Дж/м2. Величины энергетической поправки на растворение плавленого кварца составляют 0,150 Дж/м2 для раствора KOH и 0,159 Дж/м2 для LiOH. Сравнение этих значений с величинами теплот смачивания плавленого кварца (0,317 Дж/м2 в растворе KOH и 0,338 Дж/м2 в растворе LiOH) показывает, что в этом случае величина поправки достигает ~50% от измеряемой величины и с учетом этой поправки теплота смачивания может достигать величины в 0,5 Дж/м2. Необходимо подчеркнуть, однако, что растворение кислотного оксида в щелочной среде должно сопровождаться выделением значительных теплот нейтрализации. Таким образом, очевидно, что в щелочных растворах при измерении теплового эффекта взаимодействия поверхности твердого тела с раствором значительную долю могут составлять объемные, а не поверхностные тепловые эффекты. Выделить с достаточной точностью поверхностные эффекты смачивания пока не представляется возможным, Можно отметить только, что и в щелочных растворах влияние катиона качественно остается прежним.
При использовании экспериментальной теплоты смачивания в качестве характеристики разницы энтальпий границ раздела твердое тело — жидкость и твердое тело — вакуум [310] нужно помнить, что такое применение этого эффекта является оправданным только при условии неизменности объемных свойств взаимодействующих фаз. На практике, как известно, определение теплот смачивания проводится посредством контакта поверхности твердого тела с определенным объемом жидкости в измерительной ячейке калориметра. При этом некоторое количество жидкости переходит в поверхностный слой, т.е. изменяет свое состояние.
Таким образом, по способу экспериментального определения теплота смачивания представляет избыточную величину, характеризующую изменение межмолекулярных взаимодействий в граничном слое, в частности, по отношению к жидкой фазе — усиление этих взаимодействий на поверхности по сравнению с объемом. Эти изменения происходят в слое некоторой толщины. Если изменения, происходящие в системе твердое тело – жидкость, затрагивают не только поверхностный слой, но и объемы твердого тела и жидкости, то эти изменения должны учитываться при расчете энтальпии смачивания из экспериментальных данных.
Таким образом, при измерении теплот смачивания следует контролировать состав жидкой фазы после взаимодействия.
Обнаруженная специфика распределения KCl и LiCl между объемом раствора и граничным слоем и ее зависимость как от концентрации, так и от рН раствора свидетельствуют о сложности процессов, происходящих при смачивании поверхности водными растворами электролитов и о зависимости свойств граничных слоев на поверхности кварца от состава раствора.
Нужно отметить также, что, судя по одинаковому изменению теплот смачивания при разных рН при добавлении хлоридов щелочных металлов (рис. 46), отмеченная специфика распределения KCl и LiCl между граничным слоем и объемом раствора при различных рН (рис. 49) не сказывается существенно на характере изменения теплот смачивания при добавлении солей и, следовательно, не может быть использована для объяснения причин этого эффекта.
Для объяснения зависимости теплот смачивания от концентрации и типа электролита в растворе необходимы дальнейшие исследования.
IV.3.3. Оценка изменения энтропии границы кварц — раствор при
изменении состава раствора по данным теплот
и углов смачивания
Приведенные выше данные по изменению углов смачивания при изменении состава раствора, в соответствии с уравнением Юнга, позволяют оценить изменение удельной свободной энергии границы твердое тело — жидкость (Δ σ SL). Если при измерении теплот смачивания отсутствуют изменения во взаимодействующих фазах, или их можно точно учесть в суммарном тепловом эффекте, то по изменению теплоты смачивания при изменении состава раствора можно оценить изменение энтальпии границы раздела твердое тело — жидкость (Δ H SL). Тогда можно найти и изменение энтропии граничного слоя при изменении состава раствора Δ S SL по известному соотношению:
Δ G = Δ H – TΔS (IV-6)
т.е. 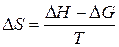 (IV-7)
(IV-7)
Проведенные нами аналитические исследования по изменению состава раствора после взаимодействия c твердой фазой показали, что при соотношении фаз, используемом при калориметрических измерениях для растворов с рН 2-6 изменений состава практически не происходит (имеется в виду содержание ионов Cl– и щелочных металлов), а тепловой эффект растворения кварца был нами оценен. Вследствие этого по полученным данным можно провести оценку изменения энтальпии границы кварц —раствор при изменении состава раствора для рН 2-6. По соотношению (IV-7) из данных по зависимости углов смачивания (рис. 27) и теплот смачивания (рис. 46,47) был проведен расчет изменения энтропии границы кварц — раствор 10-2н. HCl при появлении различного солевого фона (Δ S s), а также изменения энтропии поверхности в результате диссоциации (Δ S d) в присутствии разных солей. В качестве примера приведем расчет изменения энтропии поверхности в результате диссоциации силанольных групп (при изменении рН с 2 до 6) в присутствии KCl для кристаллического кварца:
Δ H d ≈ –Δ Q d = Q pH2 – Q pH6 = 0,391 – 0,354 = 0,037Дж/м2
Δ G d = σ LA (cos θ pH2 – cos θ pH6) = – 0,72·10-3Дж/м2
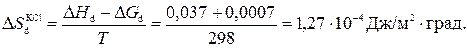
В таблице 10 представлены рассчитанные значения изменений энтропии для обоих изученных образцов кварца.
В третьих строках таблицы для каждого образца кварца приведены также величины изменения Δ S ds при изменении солевого фона:
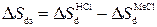 .
.
Величина Δ S ds показывает изменение энтропии процесса, связанного с диссоциацией силанольных групп при появлении в системе солевого фона 10-2н. хлоридов металлов. Как видно из таблицы, величины Δ S d во всех случаях положительны. Величины Δ S s и Δ S ds для одного и того же щелочного металла имеют одинаковые знаки, при этом знак Δ S s и Δ S ds согласуется с действием данного щелочного металла на структуру воды. На рис. 53 приведены зависимости Δ S s и Δ S ds от радиуса иона щелочных металлов. Из рис. 53 хорошо видно, что зависимости Δ S s и Δ S ds от радиуса иона имеют такой же вид, как и для растворов соответствующих солей [312,313].
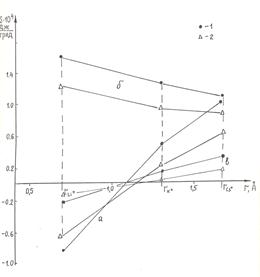 Рис. 53. Изменение энтропии поверхности кварца при изменении состава раствора в зависимости от радиуса катиона: а – в ТНЗ кварца при появлении в растворе хлоридов соответствующих металлов
Рис. 53. Изменение энтропии поверхности кварца при изменении состава раствора в зависимости от радиуса катиона: а – в ТНЗ кварца при появлении в растворе хлоридов соответствующих металлов
(10-2н.), б – при изменении pH раствора от 2 до 6 в присутствии солей, в – изменение величины ΔSds при появлении в растворе солей. 1 – кварц, 2 – КСГ.
Проведенная оценка показывает, что появление ионов щелочных металлов в граничном слое сказывается аналогично тому, как эти же ионы действуют на структуру раствора. Очень интересна, на наш взгляд, зависимость Δ S ds от вида иона. Поскольку, по электрокинетическим данным, ζ-потенциал (а следовательно, состояние ДЭС) для хлоридов щелочных металлов практически не зависит от вида металла, можно предположить, что динамика величины Δ S ds характеризует тот факт, что структура граничного слоя претерпевает изменение при изменении рН, что согласуется с данными и выводами Чернобережского с сотр. [362] и Матиевича с сотр. [371].
Таблица 10
Величины Δ S s (pH 2), Δ S d и Δ S ds кристаллического и плавленного
кварца в 10-2н. растворах солей.
| α-кварц | |||||
| 10-2н. | KCl | LiCl | CsCl | MgCl2 | BaCl2 |
| Δ S d<
Подборка статей по вашей теме:
 8213 8213 8054 8054 |