2015-06-10
2015-06-10 448
448Это наиболее часто встречающийся тип болезни. В большинстве случаев болезнь выявляется в возрасте до 30 лет и имеет постепенное начало. Течение хроническое. Диагноз может быть впервые установлен в пожилом возрасте.
Клиническая картина разнообразна и проявляется необъяснимой гепатоспленомегалией (особенно у детей), спонтанными переломами костей или болями в костях и лихорадкой. Возможны также геморрагический диатез и неспецифическая анемия.
К клиническим признакам заболевания относят также пигментацию, которая может быть диффузной или очаговой; при этом кожа имеет рыжевато-коричневый цвет. На нижних конечностях может быть симметричная свинцово-серая пигментация за счёт отложения меланина. На конъюнктиве выявляются жёлтые пингвекулы (рис. 4-10).

Рис. 4-10. Болезнь Гоше. По обеим сторонам от зрачка имеются клиновидной формы пингвекулы в виде помутнений, похожих на жир.
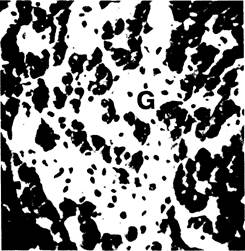
Рис. 4-11. Болезнь Гоше. В гистологических препаратах печени между тяжами гепатоцитов видны участки, заполненные крупными бледными клетками (G) с маленькими тёмными ядрами. Окраска кармином по Бесту, х250.
Селезёнка огромных размеров, печень умеренно увеличена, гладкая и плотная. Поверхностные лимфатические узлы обычно не поражаются.
Поражение печени часто сопровождается фиброзом и нарушением функциональных печёночных проб. Активность ЩФ часто повышена, иногда возрастает активность трансаминаз [23]. Могут развиться цирроз [43] и асцит. Портальная гипертензия нередко осложняется кровотечением из варикозно-расширенных вен пищевода [1].
Рентгенография костей. Длинные трубчатые кости, особенно дистальные отделы бедренной кости, расширены настолько, что имеющееся в норме сужение в надмыщелковой области исчезает. Картина при этом напоминает колбочку Эрленмейера.
В мазках костного мозга можно видеть клетки Гоше, имеющие диагностическое значение (см. рис. 4-9).
Аспирационную биопсию печени следует производить при отрицательных результатах стернальной пункции. Поражение печени носит диффузный характер (рис. 4-11).
Изменения периферической крови. При диффузном поражении костного мозга отмечается лейко-эритробластическая картина. Напротив, лейкопения и тромбоцитопения с увеличением времени кровотечения могут сопровождаться лишь умеренной гипохромной микроцитарной анемией [40].
Диагноз устанавливают на основании определения активности b-глюкоцереброзидазы в смеси мононуклеарных клеток, получаемых из венозной крови.
Изменения биохимических показателей. Активность ЩФ часто повышена. Иногда возрастает активность трансаминаз [231. Уровень сывороточного холестерина нормальный.
ЛЕЧЕНИЕ
Раньше специфической терапии этой болезни не существовало. Однако в последнее время была доказана клиническая эффективность внутривенных введений модифицированной плацентарной глюкоцереброзидазы, легли козилированной для избирательного захвата маннозовым лецитином на макрофагах. При этом отмечаются уменьшение размеров селезёнки и печени и улучшение гематологических показателей. Клинического эффекта удалось добиться, используя меньшие дозы, чем применялись ранее [6, 34], что позволяет снизить стоимость лечения.
При чрезвычайно больших размерах селезёнки, а в ряде случаев при тромбоцитопении или приобретённой гемолитической анемии производят спленэктомию или резекцию селезёнки. Полное удаление селезёнки приводит к более агрессивному поражению костей и повышению риска возникновения злокачественных опухолей [14]. В будущем успешная заместительная ферментная терапия устранит необходимость хирургического вмешательства.
При декомпенсированном циррозе производят трансплантацию печени [43|. Она не устраняет метаболический дефект, и для оценки степени повторного накопления липидов печени необходимо длительное наблюдение. Производят также ТКМ, однако её риск существенно выше, чем при проведении заместительной ферментной терапии.
Острая форма у младенцев (тип 2)
Острая форма болезни проявляется в первые 6 мес жизни. Дети обычно умирают, не достигнув 2-летнего возраста. При рождении ребёнок выглядит здоровым. Затем развиваются поражение головного мозга, прогрессирующая кахексия и нарушение психического развития. Увеличиваются размеры печени и селезёнки, могут пальпироваться также поверхностные лимфатические узлы.
При аутопсии выявляют клетки Гоше в ретикулоэндотелиальной системе. Однако они не обнаруживаются в головном мозге, и патогенез его поражения остаётся неясным.
Болезнь Ниманна-Пика
Это редкое семейное заболевание наследуется по аутосомно-рецессивному типу и встречается главным образом у евреев. Болезнь обусловлена дефицитом фермента сфингомиелиназы в лизосомах клеток ретикулоэндотелиальной системы, что приводит к накоплению сфингомиелина в лизосомах. Преимущественно поражаются печень и селезёнка.
Клетка имеет характерный вид: бледная, овальной или округлой формы, диаметром 20—40 мкм. В нефиксированном состоянии в ней видны гранулы; при фиксации жировыми растворителями гранулы растворяются, придавая тем самым клетке вакуолизированный и пенистый вид. Обычно имеется только одно или два ядра. При электронно-микроскопическом исследовании лизосомы видны как пластинчатые миелиноподобные образования. Они содержат аномальные липиды.
Болезнь Нимана—Пика типа А (острая нейронопатическая форма) встречается у детей, которые погибают, не достигнув 2-летнего возраста. Заболевание начинается в первые 3 мес жизни и проявляется анорексией, уменьшением массы тела и задержкой роста. Печень и селезёнка увеличены, кожа становится восковидной и приобретает жёлто-коричневую окраску на открытых частях тела. Поверхностные лимфатические узлы увеличены. В лёгких возникают инфильтраты. Отмечаются слепота, глухота и психические нарушения.
На глазном дне выявляют вишнёво-красные пятна, появляющиеся вследствие дегенерации сетчатки в области макулы.
Анализ периферической крови выявляет микроцитарную анемию, а на более поздних стадиях могут обнаруживаться пенистые клетки Ниманна—Пика.
Заболевание может впервые проявиться перемежающееся холестатической желтухой новорождённых. По мере развития ребёнка появляются неврологические расстройства [50].
Болезнь Ниманна—Пика типа В (хроническая форма, протекающая без поражения нервной системы) проявляется холестазом новорождённых, который разрешается спонтанно. Цирроз развивается постепенно и может привести к развитию портальной гипертензии, асцита и печёночной недостаточности [37]. Описаны случаи успешной трансплантации печени, произведённой в связи с печёночной недостаточностью [43]. Хотя на протяжении 10-месячного наблюдения не было выявлено никаких признаков отложения липидов в печени, для оценки результата в отношении метаболических нарушений требуется более длительное время.
Диагноз устанавливается на основании пункции костного мозга, которая выявляет характерные клетки Ниманна—Пика, или на основании сниженного уровня сфингомиелиназы в лейкоцитах.
Трансплантация костного мозга производилась у больных с ранним проявлением тяжёлого поражения печени [48]; предварительные результаты оказались обещающими. Отмечено снижение содержания сфингомиелина в печени, селезёнке и костном мозге.