2015-07-14
2015-07-14 5037
5037В основі синтезу пептидів лежить процес утворення пептидного (амідного) зв'язку між карбоксильною групою одної α-амінокислоти та аміногрупою — іншої. Спрощено цей процес можна подати такою схемою:
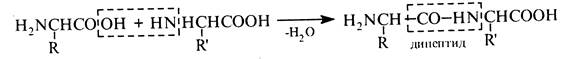
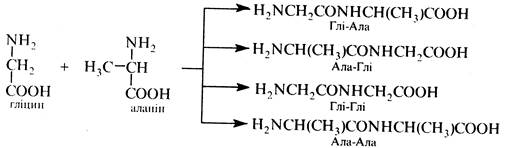
Проте, через неполярну природу α-амінокислот (цвіттер-іонна структура) проведення реакції потребує дуже високих температур, що сприяє різним небажаним побічним процесам, наприклад, циклізації з утворенням дикетопіперазинів (див. кн. 2, с 450) та ін. Крім того, в процесі синтезу виникають складнощі, пов'язані з необхідністю сполучати залишки α-амінокислот у певній послідовності. Наприклад, при взаємодії гліцину і аланіну можливе утворення чотирьох дипептидів:
У зв'язку з цим для проведення цілеспрямованого синтезу слід створити такі умови, за котрих одна з амінокислот взаємодіяла б своєю карбоксильною групою, а інша— аміногрупою. З цією метою здійснюють захист функціональних груп (-NH2 та -СООН), які не беруть участі в утворенні пептидного зв'язку. Захисні групи обирають таким чином, щоб потім кожну з них незалежно одна від одної можна було б легко видалити, не руйнуючи при цьому пептидного зв'язку.
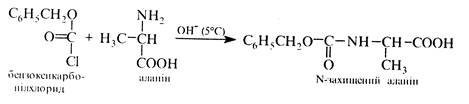
Для захисту аміногруп використовують реакцію ацилювання. найчастіше бензилоксикарбонілхлоридом або трет -бутоксикарбоксазидом. Важливою властивістю карбобензокси- та трет- бутоксикарбонільних груп є те, що вони надійно захищають хіральний центр амінокислот від рацемізалдії. Карбобензоксигрупу видаляють каталітичним гідрогенолізом, а трет -бутоксикарбонільну – за допомогою трифтороцтової кислоти. Для захисту карбоксильної групи використовують реакцію етерифікації.
Крім того, з метою підвищення ефективності процесу амідування здійснюють активацію карбоксильної групи N-захищеної амінокислоти шляхом перетворення її на хлорангідрид або на змішаний ангідрид (найчастіше взаємодією з етилхлорформіатом). Нижче наведено схему синтезу дипептиду аланіл-гліцину.
1. Захист аміногрупи аланіну
2. Активація карбоксильної групи N-захищеного аланіну
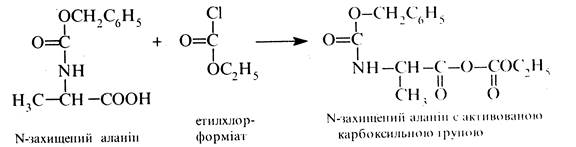
3. Захист карбоксильної групи гліцину
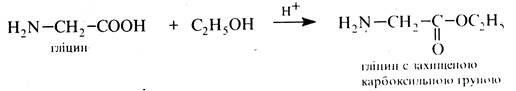
4. Утворення пептидного зв'язку та зняття захисту
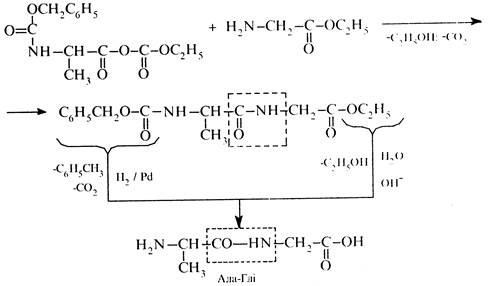
Синтез пептидів за наведеною схемою досить складний та трудоємкий.
У 1962 р. Б.Меррифілдом був запропонований більш досконалий метод добування пептидів, так званий твердофазний синтез. Суть останнього полягає в тому, що поліпептидний ланцюг оточується на твердому носії без виділення проміжних продуктів синтезу. Пептид. фіксований на носії, після кожної стадії ретельно відмивають від надлишку реагентів і побічних продуктів. Відщеплюють кінцевий продукт від носія за допомогою суміші бромоводневої та трифтороцтової кислот.
В якості твердого носія використовують зерна полімерної смоли, що містить хлорметильні (-СН2Сl) групи, котрі називають якірними групами, з якими реагує карбоксильна група N- захищеної α-амінокислоти. В результаті взаємодії відбувається фіксація С-кінця майбутнього поліпептиду на поверхні носія. Аміногрупу, як правило, захищають трет -бутоксикарбонільною групою (БОК), яка легко видаляється дією трифтороцтової кислоти. Пептидний зв'язок утворюється в присутності активатора карбоксильної групи – N,N'-дициклогексил-карбодііміду (ДЦГК) C6H11-N=C=N-C6H11. Широке застосування цієї речовини пов'язане з легкістю добування, простотою застосування, а також швидкістю та ефективністю проходження реакції конденсації в його присутності.
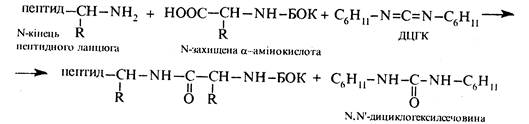
Тепер твердофазний синтез пептидів проводять у спеціальних синтезаторах, де всі етапи здійснюються автоматично з запрограмованою подачею відповідних ot-амінокислот.