2014-02-12
2014-02-12 405
405Расщепление происходит таким образом, что средняя энергия 5 d-орбиталей остается равной их средней энергии в сферическом поле. Величина расщепления обозначается Δ. Сохранение средней энергии сферического поля требует, чтобы 2 орбитали lg повышались, каждаяна 3/5 Δ, а три орбитали t2g понижались на 2/5 Δ. Величина Δ зависит от координации комплекса и степени взаимодействия лиганда с d – орбиталями.
Заполнение нижних d – орбиталей уменьшает потенциальную энергию на величину, названную энергией стабилизации кристаллическим полем (ЭСКП). Величина ЭСКП будет зависеть от размещения электронов по уровням, а характер размещения е- будет зависеть от силы поля, создаваемого лигандами.
Рассмотрим, например, два октаэдрических комплекса Fe3+ в слабом поле лигандов величина Δ будет мала, е- энергетически выгоднее разместиться по одному на каждой из d – орбиталей, в результате образуются высокоспиновые комплексы или комплексы слабого поля.
[Fe(H2O)6]3- d5, 8 группа.
— — Fe+3
— — —
|
|
|
Между е- с одинаковым спином возникает обменное взаимодействие, которое компенсирует энергию, необходимую для размещения электронов на орбиталях 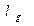 . В сильном поле величина Δ настолько велика, что этого не наблюдается, поэтому для системы энергетически выгоднее размещение электронов на уровнях
. В сильном поле величина Δ настолько велика, что этого не наблюдается, поэтому для системы энергетически выгоднее размещение электронов на уровнях 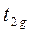 . При этом образуются низкоспиновые комплексы или комплексы сильного поля.
. При этом образуются низкоспиновые комплексы или комплексы сильного поля.
[Fe(CN)6]4- d6, 8 группа.
— — Fe+2
— — —
C учетом образования низкоспиновых и высокоспиновых комплексов и сосчитав все распределения электронов на орбиталях и можно оценить энергию стабилизации кристаллическим полем.
Теория поля лигандов, развита на основе метода молекулярных орбиталей, является точным приближением по сравнению с теорией кристаллического поля. В отличие от теории кристаллического поля она учитывает электронную структуру не только центрального атома, но и лигандов. Применение теории поля лигандов не изменяет представлений о расщеплении d - орбиталей в электростатическом поле и образовании низкоспиновых и высокоспиновых комплексов. Теория поля лигандов позволяет предсказать, какие атомные орбитали центрального иона пригодны для образования связей с лигандами, какова степень ковалентности или полярности возникающей связи. На основе теории поля лигандов возникли представления о реализации π – комплексного и т. механизма в реакциях гидрирования и окисления.
π – комплексный механизм – за счет е- 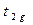 зоны с разрыхляющей π* орбиталью, например, С2Н4 σ – комплекс – в результате адсорбции протона связь осуществляется за счет свободной – зоны и е- s – орбиталей.
зоны с разрыхляющей π* орбиталью, например, С2Н4 σ – комплекс – в результате адсорбции протона связь осуществляется за счет свободной – зоны и е- s – орбиталей.
|
|
|
С этих позиций активация молекул в каталитических реакциях определяется образованием комплексов в результате их присоединения к отдельным ионам катализатора. При таком подходе хемосорбционная связь и активация молекул локальны, а макроскопические коллективные свойства играют второстепенную роль. Хемосорбция на катионе переходного металла подобна реакции комплексобразования с увеличением числа лигандов, причем одним из лигандов будет адсорбированная молекула, а остальными – ионы кристаллической решетки.
При такой адсорбции происходит увеличение координационного числа, что повышает энергию стабилизации кристаллическим полем.
Рассмотрим адсорбцию O2 на NiO - чисто ионный кристалл.
O-2 Ni+2 O-2 Ni+2 O-2 Ni+2
+ O2 →
Ni+2 O-2 Ni+2 O-2 Ni+2 O-2
O2 → 2O. NiO – квадратная пирамида
Теплота адсорбции q будет определяться по уравнению:
q = -1/2D – J + Z + W’ + K (ЭСКП)
в расчете на 1 атом,
D – энергия связи в молекуле О2 (энергия диссоциации),
J – потенциал ионизации,
Z – сродство к электрону,
W’ – энергия взаимодействия,
К – энергия стабилизации кристаллическим полем.
Дауден и Уэллс впервые в 1960 г. сопоставили каталитические свойства с величиной К (ЭСКП). Была обнаружена зависимость:
Е = ± α q.
При сопоставлении каталитических свойств окислов IV периода с величиной К (ЭСКП) Дауденом была обнаружена следующая двухпиковая зависимость:

TiO2 V2O5 V2O3 Cr2O3 MnO Fe2O3 Co3O4 NiO Cu2O CuO ZnO
При изменении природы катализатора, главную роль вносит К (ЭСКП), то теория ограничивается рамками соотношения Бренстеда – Поляни. Если рассчитать W и соотнести с уравнение Бренстеда –Поляни, то можно провести корреляцию между W и каталитической активностью.
Изменение К (ЭСКП) аналогично. Обнаружено для реакции гидрирования олефинов, окисления СО, Н2, Н2 –D2 – обмена.
Изменение каталитической активности совпадает с изменением величины К (ЭСКП).
Первый максимум на графике – конфигурация атома d3, второй максимум – d6d7.
Минимум – d5, d10, Co, Cu2O, ZnO, CaO, TiO2 (по цвету).
Эта зависимость более закономерна, чем предполагалось, впоследствии это подтвердилось для большого числа реакций.
Эти данные подтверждают роль локального взаимодействия в гетерогенном катализе. Вероятность локальных взаимодействий подтверждена и при исследовании каталитических реакций на разбавленных твердых растворах ионов переходных металлов в твердых матрицах. Чтобы ослабить коллективное взаимодействие между ионами переходных металлов, адсорбцию и катализ изучали на ионах переходных металлов, разведенных в твердой матрице.
Так, например, оказалось удобным разбавить окисел металла типа МеО в MgO, неактивной в исследуемой реакции, для окислов типа Ме2О3 можно использовать в качестве матрицы Al2O3. Для ряда реакций наблюдается рост атомной каталитической активности (активности на один атом или ион) в таких областях разведения, когда коллективное взаимодействие практически отсутствует. Это показывает, что перенос электронов по зонам проводимости не всегда обязателен для катализа и более важны свойства индивидуального окисла.
Такой подход к явлениям адсорбции и катализа позволяет объяснить:
1. сходство в каталитическом действии одних и тех же элементов в виде простых твердых тел (Ме), в виде их твердых соединений (окислы, сульфиды) и в виде сольватированных ионов в растворе.
2. роль хемосорбции в катализе.
3. особое место переходных элементов в катализе без использования спорной модели d – зоны.
Такой подход тоже односторонен, учитывает только одно слагаемое, а это не позволяет предсказать, в каких случаях такая зависимость будет наблюдаться и каково электронное строение катиона, отвечающее оптимальному катализатору.