 2015-06-14
2015-06-14 2053
2053ФЕДЕРАЛЬНОЕ АГЕНСТВО ПО КУЛЬТУРЕ И КИНЕМАТОГРАФИИ
Государственное образовательное учреждение высшего
профессионального образования
"САНКТ-ПЕТЕРБУРГСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ КИНО И ТЕЛЕВИДЕНИЯ"
Кафедра общей, органической и физической химии
ФИЗИЧЕСКАЯ ХИМИЯ
Методические указания по выполнению лабораторных
работ для студентов специальности 240504.
"Технология кинофотоматериалов и магнитных носителей"
дневного и заочного отделений.
Часть V. Поверхностные явления.
Санкт-Петербург
Составитель: Л.Л. Кузнецов
Рецензент: В.В. Митрофанов
Рекомендовано к изданию в качестве методических указаний по лабораторному практикуму по физической химии кафедрой общей, органической и физической химии.
Протокол заседания кафедры общей, органической и физической химии № 3 от 8 декабря 2005 г.
ã СПбГУКиТ 2006 г.
1. Межмолекулярное взаимодействие
Валентно-насыщенные молекулы, такие, например, как Н2O, СН4, NН3, СO2 и т.д., взаимодействуют между собой, о чем свидетельствует возможность существования их в жидком и твердом состояниях. Испарение жидкостей всегда сопровождается поглощением тепла, что связано с разрушением "связей" между отдельными молекулами. Величина мольной теплоты испарения является мерой энергии межмолекулярного взаимодействия в жидкости (чем больше энергия взаимодействия, тем больше теплота испарения).
| Жид-кость | Теплота испарения, кДж/моль |
| CH4 | 8.2 |
| C3H8 | 18.6 |
| HJ | 19.8 |
| NH3 | 23.3 |
| C2H5OH | 38.6 |
| H2O | 40.7 |
Из приведенной таблицы видно, что с ростом полярности молекул энергия их межмолекулярного взаимодействия растет, однако даже у абсолютно неполярных молекул (СН4, С3Н8) она не равна нулю. Основные виды взаимодействия между частицами делятся на: неспецифические (ван-дер-ваальсово взаимодействие, ион-дипольное взаимодействие) и специфические (водородная связь).
1.1. Ван-дер-ваальсово взаимодействие.
Оно осуществляется на сравнительно больших расстояниях, до 10∙10-10 м и имеет электростатическую природу. Основными составляющими взаимодействия являются: дисперсионное, индукционное, ориентационное и ион-дипольное.
1.1.1. Дисперсионное взаимодействие.
Такое взаимодействие осуществляется между любыми молекулами, как полярными, так и неполярными и заключается в том, что при сближении молекул на расстояние 3−5∙10-10 м колебания их электронных оболочек, происходящие примерно 1015 раз в секунду, становятся согласованными. Мгновенно образующиеся диполи ориентируются друг относительно друга и меняются "в такт". Это приводит к понижению общей энергии системы из двух молекул, то есть к их притяжению.
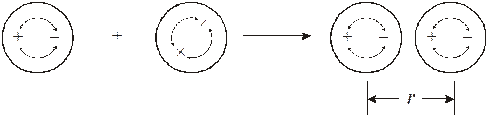
Для неполярных молекул дисперсионное взаимодействие является единственным видом межмолекулярного взаимодействия. Оно является сравнительно дальнодействующим и его энергия убывает пропорционально шестой степени расстояния между молекулами:
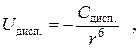
где  − коэффициент пропорциональности, зависящий от структуры частиц.
− коэффициент пропорциональности, зависящий от структуры частиц.
Отрицательное значение энергии (знак минус) показывает, что это энергия притяжения.
1.1.2. Индукционное взаимодействие.
Оно проявляется в том случае, когда хотя бы одна из взаимодействующих молекул является полярной, то есть представляет собой постоянный диполь. При взаимодействии неполярной молекулы с соседней полярной молекулой происходит деформация электронной оболочки и образование наведенного, или индуцированного диполя. Результатом этого является электростатическое притяжение двух диполей:
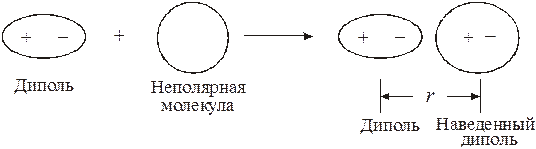
Энергия индукционного взаимодействия тем больше, чем больше дипольный момент полярной молекулы (диполя) и поляризуемость электронной оболочки неполярной молекулы. Эта энергия убывает также пропорционально шестой степени расстояния между частицами.
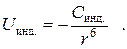
1.1.3. Ориентационное взаимодействие.
Этот вид взаимодействия проявляется лишь в том случае, когда обе взаимодействующие молекулы являются полярными. При сближении таких молекул происходит их ориентация либо "голова к хвосту", либо "боками". Такая ориентация приводит к наибольшему уменьшению энергии и, соответственно, к максимальному притяжению. Реализуется она лишь в молекулярных кристаллах (в кристаллах льда, например). В жидкостях тепловое движение несколько "разбрасывает", или "разориентирует" диполи, однако и в этом случае ориентация диполей имеет место, поскольку является энергетически выгодной.
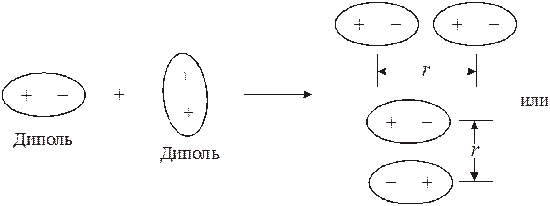
Энергия ориентационного взаимодействия убывает пропорционально шестой степени расстояния между молекулами.
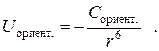
Таким образом, энергия ван-дер-ваальсова взаимодействия складывается из перечисленных видов взаимодействия. Вклад того или иного вида взаимодействия зависит от строения молекул. Чем более полярны взаимодействующие молекулы, тем большую долю имеет ориентационное, или диполь-дипольное взаимодействие. Чем менее полярны – тем больше доля дисперсионного взаимодействия. Для совершенно неполярных молекул его доля составляет, естественно, 100 %. С увеличением полярности молекулы суммарная энергия ван-дер-ваальсова взаимодействия возрастает. Чем меньше расстояние между молекулами, тем его энергия также выше:
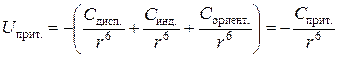
 .
.
Если бы между молекулами жидкости, твердых веществ и газов существовали только силы притяжения, они сближались бы на нулевое расстояние, в результате чего плотность вещества возрастала бы почти неограниченно. Тот факт, что плотность жидких и кристаллических тел имеет вполне определенную величину, указывает на одновременное существование сил межмолекулярного отталкивания. Эти силы имеют квантово-механическую природу и связаны с принципом Паули, запрещающим перекрывание электронных оболочек.
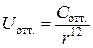
Их действие проявляется на очень небольших расстояниях. Энергия отталкивания убывает пропорционально 12 степени расстояния. Общая энергия взаимодействия двух молекул складывается из энергии ван-дер-ваальсова притяжения и отталкивания:
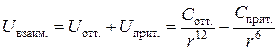 .
.
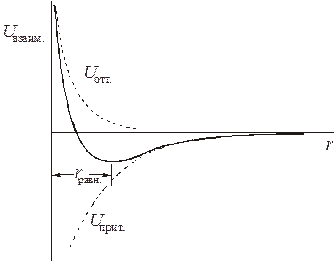 Зависимость
Зависимость 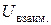 от расстояния между молекулами является экстремальной. Молекулы в жидкостях и кристаллах располагаются на расстояниях
от расстояния между молекулами является экстремальной. Молекулы в жидкостях и кристаллах располагаются на расстояниях 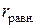 , соответствующих минимуму энергии. Равновесное расстояние между молекулами обычно составляет 3−5∙10-10 м, что в несколько раз превышает обычную длину химической связи, а энергия ван-дер-ваальсова взаимодействия (
, соответствующих минимуму энергии. Равновесное расстояние между молекулами обычно составляет 3−5∙10-10 м, что в несколько раз превышает обычную длину химической связи, а энергия ван-дер-ваальсова взаимодействия (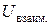 ) редко превышает ~5 кДж/моль, что примерно на два порядка меньше энергии химической связи.
) редко превышает ~5 кДж/моль, что примерно на два порядка меньше энергии химической связи.
1.2. Ион – дипольное взаимодействие.
Оно проявляется в случае, когда одна из взаимодействующих частиц является ионом, а другая диполем, как в случае раствора электролита в полярном растворителе (вода, спирты, аммиак и т.д.). Энергия взаимодействия этого вида тем выше, чем больше заряд иона, а также дипольный момент молекулы и ее поляризуемость, поскольку основным в этом случае является электростатическое (кулоновское) притяжение и индукционное взаимодействие. Энергия кулоновского притяжения убывает пропорционально квадрату, а энергия индукционного взаимодействия между ионом и диполем – четвертой степени расстояния:
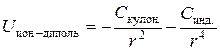 .
.
Силы притяжения между ионом и диполем являются более дальнодействующими по сравнению с притяжением незаряженных частиц. Они уравновешиваются силами отталкивания, аналогичными проявляющимся в межмолекулярном взаимодействии. Между ионом и диполем устанавливается равновесное расстояние (3−5∙10-10 м). Энергия такого взаимодействия в несколько раз выше энергии межмолекулярного взаимодействия и для системы однозарядный ион – молекула воды несколько превышает 40 кДж/моль.
1.3. Водородная связь.
Такая связь возникает между акцептором электронов – молекулой имеющей в своем составе поляризованную связь атома водорода с каким либо электроотрицательным атомом (А←Н) и донором электронов – молекулой (В), в составе которой имеются атомы с заполненными несвязывающими n –орбиталями (свободными электронными парами), такие как:
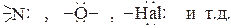
При сближении донора и акцептора на расстояние, сравнимое с длиной обычной химической связи (1.5−3∙10-10 м), происходит образование трехцентровой водородной связи, обусловленной образованием трех молекулярных орбиталей из двух σ–орбиталей акцептора (связывающей и разрыхляющей) и одной несвязывающей n –орбитали донора. Ее прочность обычно находится в пределах от 15−20 кДж/моль до ~200 кДж/моль, то есть от энергии, сравнимой с энергией ион-дипольного взаимодействия, до энергии, близкой к энергии обычной химической связи. В качестве примера можно привести образование водородной связи в дифторид-анионе:
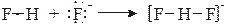 .
.
Ее прочность практически не отличается от прочности обычной химической связи F−H. Сравнительно высокая прочность водородной связи является причиной ассоциации многих молекул в жидком и даже в газообразном состояниях. Так, молекулы карбоновых кислот в органических растворителях димеризованы нацело, а в парах – частично:
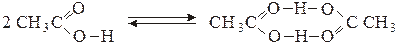
Молекулы воды и спиртов в жидком состоянии и даже в парах также в значительной степени ассоциированы:
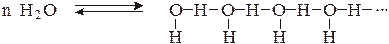
Результатом этого является значительное увеличение энергии, требующейся для испарения одного моля жидкости, а также увеличение самой температуры кипения по сравнению с температурами кипения неассоциированных жидкостей с близкой молярной массой. Так, если бы вода не была ассоциированной жидкостью, ее температура кипения была бы не +100ºС, а близкой к температуре кипения метана (–161ºС).
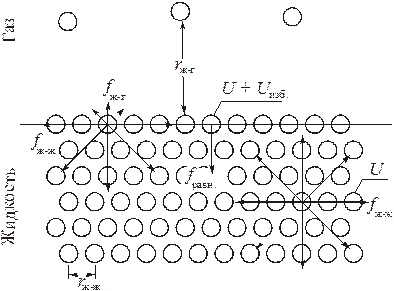 Если рассмотреть гетерогенную систему, состоящую из двух фаз, разделенных поверхностью раздела, можно заметить, что свойства частиц вещества (молекул, ионов) вблизи поверхности раздела должны заметно отличаться от их свойств в глубине фазы.
Если рассмотреть гетерогенную систему, состоящую из двух фаз, разделенных поверхностью раздела, можно заметить, что свойства частиц вещества (молекул, ионов) вблизи поверхности раздела должны заметно отличаться от их свойств в глубине фазы.
В качестве примера рассмотрим поверхность раздела "жидкость – газ". Любая молекула жидкости, находящаяся внутри фазы и окруженная со всех сторон одинаковыми молекулами, взаимодействует с ними таким образом, что равнодействующая всех сил, действующих на нее, равна нулю. В то же время энергия взаимодействия поверхностной молекулы жидкости с молекулами газа (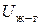 ) значительно меньше энергии взаимодействия ее с молекулами жидкости (
) значительно меньше энергии взаимодействия ее с молекулами жидкости (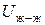 ), поскольку молекулы газа находятся от нее на значительно большем расстоянии, чем окружающие молекулы жидкости. В результате – равнодействующая сил, действующих на каждую поверхностную молекулу, уже не равна нулю и направлена вглубь жидкой фазы перпендикулярно поверхности раздела. Благодаря этому, любая поверхностная молекула жидкости кроме внутренней энергии (U), определяемой структурой молекулы и температурой, имеет и избыточную потенциальную энергию (
), поскольку молекулы газа находятся от нее на значительно большем расстоянии, чем окружающие молекулы жидкости. В результате – равнодействующая сил, действующих на каждую поверхностную молекулу, уже не равна нулю и направлена вглубь жидкой фазы перпендикулярно поверхности раздела. Благодаря этому, любая поверхностная молекула жидкости кроме внутренней энергии (U), определяемой структурой молекулы и температурой, имеет и избыточную потенциальную энергию (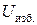 ), величина которой тем больше, чем больше равнодействующая сила, втягивающая поверхностную молекулу вглубь фазы. Таким образом, избыточная потенциальная энергия поверхностной молекулы тем больше, чем больше различие в энергиях взаимодействия молекул жидкости между собой и молекул жидкости с молекулами газа.
), величина которой тем больше, чем больше равнодействующая сила, втягивающая поверхностную молекулу вглубь фазы. Таким образом, избыточная потенциальная энергия поверхностной молекулы тем больше, чем больше различие в энергиях взаимодействия молекул жидкости между собой и молекул жидкости с молекулами газа.
Суммарная избыточная энергия молекул, находящихся на 1 м2 поверхности раздела, называется поверхностным натяжением. Оно равно, также, работе, которую необходимо совершить для образования 1 м2 новой поверхности раздела. При постоянной температуре и постоянном давлении
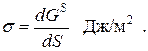
Кроме этого, поверхностное натяжение численно равно силе, действующей на единицу длины контура поверхности раздела тангенциально к поверхности. Его размерность составляет, также, Дж/м2 = Н·м/м = Н/м:
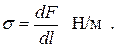
Чем больше энергия взаимодействия молекул жидкости между собой () и чем меньше энергия взаимодействия молекул жидкости с молекулами газа (), то есть чем больше разница в 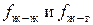 , тем больше величина
, тем больше величина 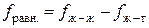 , тем больше поверхностное натяжение на поверхности раздела. По этой причине поверхностное натяжение на границе раздела "вода – воздух" значительно выше, чем на границе "гексан – воздух", но значительно меньше, чем на границе "ртуть – воздух":
, тем больше поверхностное натяжение на поверхности раздела. По этой причине поверхностное натяжение на границе раздела "вода – воздух" значительно выше, чем на границе "гексан – воздух", но значительно меньше, чем на границе "ртуть – воздух":
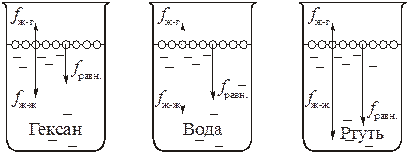
s = 0.028 < s = 0.072 < s = 0.472 Дж/м2
Причиной этого является то, что если неполярные молекулы гексана связаны лишь дисперсионными силами, то полярные молекулы воды связаны не только ван-дер-ваальсовыми силами, но и водородной связью, а атомы и ионы ртути – гораздо более прочной металлической связью.
Поскольку поверхностное натяжение зависит и от энергии взаимодействия молекул одной фазы с молекулами другой фазы, оно уменьшается в ряду: "вода – воздух" > "вода – гексан" > "вода – спирт". В последнем случае поверхностное натяжение практически равно нулю и жидкости, не разделенные поверхностью раздела, смешиваются.
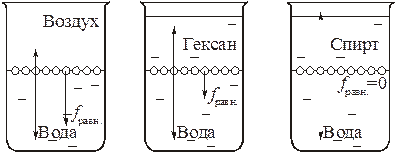
| σ > | σ > | σ = 0 |
С ростом давления газа уменьшается расстояние между молекулами газа и жидкости ( ), поэтому увеличивается энергия их взаимодействия (), а значит и
), поэтому увеличивается энергия их взаимодействия (), а значит и  . Вследствие этого уменьшается сила, втягивающая поверхностные молекулы вглубь фазы () и поверхностное натяжение уменьшается. Так, при 150 атм. величина поверхностного натяжения на поверхности "вода – воздух" вдвое меньше чем при нормальном давлении.
. Вследствие этого уменьшается сила, втягивающая поверхностные молекулы вглубь фазы () и поверхностное натяжение уменьшается. Так, при 150 атм. величина поверхностного натяжения на поверхности "вода – воздух" вдвое меньше чем при нормальном давлении.
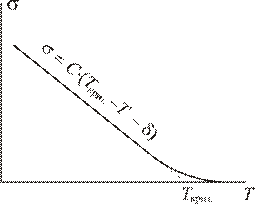 С ростом температуры увеличивается среднее расстояние между молекулами жидкости (вещества при нагревании расширяются) и, следовательно, уменьшается энергия их взаимодействия (). С другой стороны, рост температуры приводит к увеличению давления насыщенного пара жидкости и соответствующему уменьшению расстояния между молекулами жидкости и газа. Это увеличивает энергию взаимодействия поверхностных молекул с молекулами газа (). Все вместе это приводит к уменьшению поверхностного натяжения на границе "жидкость – газ". Зависимость σ от Т является линейной почти вплоть до критической температуры, при которой поверхностное натяжение уменьшается до нуля:
С ростом температуры увеличивается среднее расстояние между молекулами жидкости (вещества при нагревании расширяются) и, следовательно, уменьшается энергия их взаимодействия (). С другой стороны, рост температуры приводит к увеличению давления насыщенного пара жидкости и соответствующему уменьшению расстояния между молекулами жидкости и газа. Это увеличивает энергию взаимодействия поверхностных молекул с молекулами газа (). Все вместе это приводит к уменьшению поверхностного натяжения на границе "жидкость – газ". Зависимость σ от Т является линейной почти вплоть до критической температуры, при которой поверхностное натяжение уменьшается до нуля:
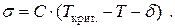
Здесь С и 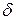 − постоянные, зависящие от свойств жидкости.
− постоянные, зависящие от свойств жидкости.
Совершенно аналогичные закономерности наблюдаются и на поверхностях твердых тел. И в этом случае поверхностное натяжение тем больше, чем больше разница в энергиях взаимодействия поверхностных молекул с молекулами своей и противоположной фазы. Поскольку плотность вещества в твердом состоянии обычно выше чем в жидком, то частицы в кристаллах располагаются ближе друг к другу. Соответственно выше энергия их взаимодействия, выше и поверхностное натяжение на границе раздела газа с твердым веществом. Особенно велико оно на поверхности ионных кристаллов.
3. Поверхностное натяжение растворов. ПАВ и ПИАВ.
Поверхностное натяжение на границе раздела "водный раствор – газ" зависит от вида и концентрации растворенного в воде вещества. По влиянию на величину поверхностного натяжения такие вещества делятся на поверхностно-активные (ПАВ) и поверхностно-инактивные вещества (ПИАВ). ПАВ уменьшают поверхностное натяжение вследствие того, что, будучи менее полярными чем молекулы воды, взаимодействуют между собой и с молекулами воды с энергией меньшей, чем молекулы воды между собой:
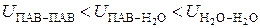 .
.
По этой причине на поверхностные молекулы ПАВ действует значительно меньшая втягивающая сила чем на молекулы воды. По этой причине малополярные молекулы ПАВ имеют меньшую избыточную энергию чем высокополярные молекулы воды. Находясь в поверхностном слое, молекулы ПАВ уменьшают суммарную избыточную энергию поверхностных молекул, то есть поверхностное натяжение.
Поверхностно-активными являются любые малополярные органические вещества, имеющие в своем составе неполярный алифатический, перфторалифатический или жирноароматический радикал:
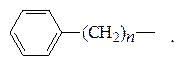
СН3–(СН2) n –СН2– CF3–(CF2) m –CF2– или
Кроме него в состав молекулы обязательно должна входить и какая либо полярная группа, обеспечивающая ПАВ некоторую растворимость в воде:
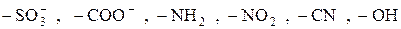 и т.д.
и т.д.
Таким образом, любая молекула ПАВ схематично может быть представлена как:
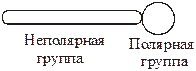
Наибольшее распространение получили так называемые ионогенные ПАВ. При их диссоциации в воде образуются поверхностно-активные ионы. В качестве примера можно привести соли жирных кислот, входящие в состав хозяйственного мыла или сульфокислот, составляющих основу стиральных порошков. Такие ПАВ называются анионоактивными, например:
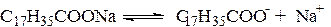
Примером катионоактивных ПАВ могут служить четвертичные соли аминов, дающие при диссоциации поверхностно-активные катионы, например:
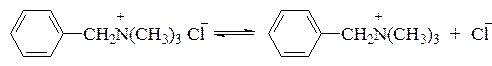
Неионогенными ПАВ являются высшие алифатические спирты, амины и т.д., например бутиловый спирт CH3CH2CH2CH2OH, амиловый спирт CH3CH2CH2CH2CH2OH.
Мерой влияния ПАВ на поверхностное натяжение раствора является величина его поверхностной активности, показывающая степень уменьшения поверхностного натяжения раствора с ростом его концентрации. В честь Гиббса поверхностная активность обозначается буквой G, измеряется в Дж·м/моль и равна производной поверхностного натяжения по концентрации ПАВ, взятой с обратным знаком:
G = – 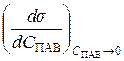 .
.
Для ПАВ эта величина всегда положительна.
Согласно правилу Траубе абсолютная величина поверхностной активности ПАВ растет приблизительно в 3.2 раза с увеличением длины цепочки алифатического радикала в его молекуле на одну метиленовую группу. Это правило соблюдается, естественно, лишь для ПАВ, принадлежащих к одному гомологическому ряду. Так, небольшие добавки бутилового спирта CH3(CH2)3OH понижают поверхностное натяжение воды примерно в 3 раза в меньшей степени, чем такие же добавки амилового спирта, то есть CH3(CH2)4OH.
Поверхностно-инактивными веществами являются такие вещества, молекулы которых взаимодействуют между собой и с молекулами воды с большей энергией чем молекулы воды между собой:
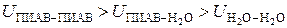
Такие молекулы втягиваются с поверхности раздела вглубь фазы с силой большей чем молекулы воды, поэтому имеют большую, по сравнению с молекулами воды, избыточную энергию. Попадая в поверхностный слой, молекулы ПИАВ увеличивают суммарную избыточную энергию поверхностных молекул. Примерами ПИАВ могут служить любые электролиты, диссоциирующие с образованием гидратированных ионов, например:
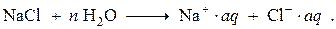
Энергия ион-дипольного взаимодействия ионов с молекулами воды и, тем более, межионного притяжения противоположно заряженных ионов, значительно превышает энергию взаимодействия молекул воды.
РАБОТА № 7
АДСОРБЦИЯ НА ТВЕРДЫХ ПОВЕРХНОСТЯХ, НАХОЖДЕНИЕ КОНСТАНТ УРАВНЕНИЯ ФРЕЙНДЛИХА ПРИ АДСОРБЦИИ ИЗ РАСТВОРОВ.
Цель работы: построение зависимости степени адсорбции карбоновой кислоты из водного раствора на активированном угле от равновесной ее концентрации, вычисление значений коэффициентов уравнения Фрейндлиха.
1. ТЕОРЕТИЧЕСКАЯ ЧАСТЬ
1.1. Адсорбция газов и паров на твердых поверхностях.
Общая энергия системы из твердой, газообразной фазы и поверхности раздела между ними при постоянной температуре и давлении определяется суммой энергий Гиббса фаз и избыточной энергии Гиббса поверхности раздела:
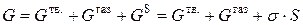 .
.
При неизменных 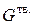 и
и 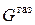 изменение энергии Гиббса системы определяется изменением либо поверхностного натяжения, либо площади поверхности раздела:
изменение энергии Гиббса системы определяется изменением либо поверхностного натяжения, либо площади поверхности раздела:
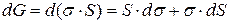 .
.
Самопроизвольными являются любые процессы, приводящие к уменьшению энергии системы (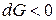 ). Площадь поверхности раздела "твердое тело – газ" есть величина постоянная (S = const, dS = 0) поэтому
). Площадь поверхности раздела "твердое тело – газ" есть величина постоянная (S = const, dS = 0) поэтому 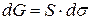 , а значит самопроизвольными в этом случае являются процессы, сопровождающиеся уменьшением поверхностного натяжения (
, а значит самопроизвольными в этом случае являются процессы, сопровождающиеся уменьшением поверхностного натяжения (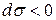 ).
).
Поверхностное натяжение на границе раздела "твердое тело – газ" определяется различием в энергии взаимодействия поверхностных молекул твердого вещества с молекулами своей фазы (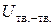 ) и с молекулами газа (
) и с молекулами газа (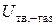 ). Это различие тем меньше, чем выше величина , определяемая расстоянием между поверхностью раздела и молекулами газа (
). Это различие тем меньше, чем выше величина , определяемая расстоянием между поверхностью раздела и молекулами газа (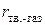 ). Поскольку при сгущении газа на поверхности раздела это расстояние уменьшается, величина растет, а поверхностное натяжение уменьшается. Соответственно уменьшается и общая энергия системы. По этой причине такой процесс протекает самопроизвольно.
). Поскольку при сгущении газа на поверхности раздела это расстояние уменьшается, величина растет, а поверхностное натяжение уменьшается. Соответственно уменьшается и общая энергия системы. По этой причине такой процесс протекает самопроизвольно.
Самопроизвольное сгущение газа или пара на поверхности твердого вещества, называется адсорбцией. Твердое тело, на котором адсорбируется газ, называется адсорбентом, а адсорбирующееся вещество – адсорбатом. Если взаимодействие молекул газа с молекулами твердого вещества ограничивается физическим, или ван-дер-ваальсовым взаимодействием (см. теоретическое введение), адсорбция называется физической. Если же взаимодействие сопровождается образованием поверхностных химических соединений, адсорбция называется химической, или хемосорбцией.
Энергия взаимодействия молекул адсорбента и адсорбата при физической адсорбции составляет единицы и очень редко, при образовании водородных связей – десятки кДж/моль. При химической адсорбции эта энергия равна энергии обычных химических связей и составляет сотни кДж/моль. Поскольку молекулы адсорбата участвуют в тепловом движении, энергия которого при нормальной температуре составляет около 4 кДж/моль, часть молекул газа, связанных с поверхностью адсорбента ван-дер-ваальсовыми силами, отрывается от нее и переходит в объем газовой фазы. Этот процесс называется десорбцией. При данных условиях между процессом адсорбции и десорбции устанавливается равновесие, характеризующееся определенной величиной равновесной степени адсорбции.
Энергии теплового движения совершенно недостаточно для разрыва химических связей, поэтому десорбция протекает лишь при физической адсорбции. Химическая адсорбция при температурах до ~103 K является необратимой.
1.2. Теория мономолекулярной адсорбции Лэнгмюра.
Согласно этой теории поверхность адсорбента является совершенно однородной. По ней равномерно распределены центры адсорбции, способные образовывать ван-дер-ваальсовы связи с молекулами адсорбата. Один центр адсорбции образует связь только с одной молекулой адсорбата, поэтому при максимальной адсорбции на поверхности адсорбента образуется мономолекулярный слой адсорбата.
Скорость адсорбции пропорциональна величине свободной, незанятой адсорбатом поверхности адсорбента и числу соударений молекул газа об эту поверхность в единицу времени. Если долю поверхности, занятой адсорбатом обозначить как 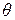 , доля свободной поверхности будет составлять (
, доля свободной поверхности будет составлять (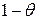 ). Число соударений пропорционально давлению газа P, поэтому скорость адсорбции составляет:
). Число соударений пропорционально давлению газа P, поэтому скорость адсорбции составляет:
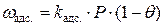
Скорость десорбции при данной температуре пропорциональна числу молекул адсорбата на единице поверхности адсорбента, то есть величине 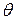 :
:
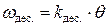
При равновесии скорости адсорбции и десорбции газа равны друг другу, то есть 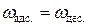 и, следовательно,
и, следовательно,
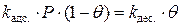 ;
; 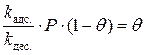 ;
; 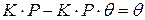 ,
,
откуда 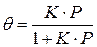 ,
,
где 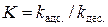 – константа равновесия адсорбции.
– константа равновесия адсорбции.
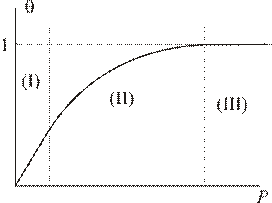 Это уравнение называется адсорбционным уравнением Лэнгмюра. Из него следует, что степень адсорбции, выражаемая величиной доли поверхности, занятой адсорбатом, в общем случае является нелинейной функцией давления. При малых давлениях газа, когда
Это уравнение называется адсорбционным уравнением Лэнгмюра. Из него следует, что степень адсорбции, выражаемая величиной доли поверхности, занятой адсорбатом, в общем случае является нелинейной функцией давления. При малых давлениях газа, когда 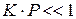 степень адсорбции линейно растет с давлением:
степень адсорбции линейно растет с давлением: 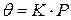 (область (I) графика). При достаточно высоких давлениях, когда
(область (I) графика). При достаточно высоких давлениях, когда 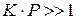 , вся поверхность адсорбента оказывается занята адсорбатом (область (III) графика) и для нее
, вся поверхность адсорбента оказывается занята адсорбатом (область (III) графика) и для нее 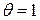 . Средняя нелинейная часть графика описывается полным уравнением Лэнгмюра.
. Средняя нелинейная часть графика описывается полным уравнением Лэнгмюра.
Уравнение Лэнгмюра показывает, что при данном давлении степень адсорбции тем больше, чем больше величина константы равновесия адсорбции 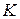 . В свою очередь, константа равновесия адсорбции зависит от изменения энергии Гиббса в процессе адсорбции и от температуры:
. В свою очередь, константа равновесия адсорбции зависит от изменения энергии Гиббса в процессе адсорбции и от температуры:
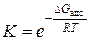
Поскольку адсорбция является самопроизвольным процессом, при котором энергия Гиббса системы уменьшается, то 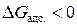 . Уменьшается также и энтропия системы (при образовании связи адсорбент-адсорбат возрастает упорядоченность системы и поэтому
. Уменьшается также и энтропия системы (при образовании связи адсорбент-адсорбат возрастает упорядоченность системы и поэтому 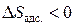 ). Из соотношения
). Из соотношения 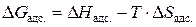 следует, что
следует, что 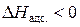 , то есть теплота при адсорбции всегда выделяется. Чем больше теплоты выделяется при адсорбции одного моля адсорбата, тем отрицательнее оказывается величина
, то есть теплота при адсорбции всегда выделяется. Чем больше теплоты выделяется при адсорбции одного моля адсорбата, тем отрицательнее оказывается величина 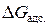 и тем больше величина константы равновесия адсорбции . В соответствии с уравнением Лэнгмюра большей оказывается и степень адсорбции.
и тем больше величина константы равновесия адсорбции . В соответствии с уравнением Лэнгмюра большей оказывается и степень адсорбции.
Чем выше температура, тем выше энергия теплового движения молекул адсорбата на поверхности адсорбента, тем чаще они отрываются от поверхности, тем, соответственно, меньше константа равновесия адсорбции, тем меньше и степень адсорбции.
При адсорбции смеси газов происходит вытеснение из адсорбционного слоя газа с меньшей теплотой адсорбции и замена его газом, теплота адсорбции которого выше.
1.3. Теория полимолекулярной адсорбции.
Мономолекулярная адсорбция газов и паров на поверхности адсорбента встречается достаточно редко. Значительно чаще изотермы адсорбции имеют более сложный вид, чем следует из теории Лэнгмюра. Для объяснения такой зависимости степени адсорбции от давления адсорбата была предложена теория полимолекулярной адсорбции, согласно которой центры адсорбции способны образовывать связи не только с одной молекулой адсорбата с образованием единичного комплекса, но и с двумя, тремя и более молекулами. При этом образуются соответственно двойные, тройные и более комплексы. Наибольшая теплота выделяется при образовании связи с первой молекулой адсорбата, значительно меньше – при образовании связи со второй, еще меньше – с третьей и т.д. Соответственно, наибольшей оказывается константа равновесия адсорбции первой молекулы, значительно меньше – второй и т.д. молекулы адсорбата. Пока поверхность адсорбента занята лишь в небольшой степени, преобладают единичные комплексы. После заполнения поверхности мономолекулярным слоем начинают образовываться двойные, тройные и т.д. комплексы с образованием второго, третьего и т.д. слоев адсорбата вплоть до конденсации жидкой пленки, что происходит при давлении насыщенного пара (P = P нас.).
1.4. Адсорбция на пористых адсорбентах.
Капиллярная конденсация. Пористые адсорбенты, такие как активированный уголь, цеолит, глины имеют значительно большую удельную поверхность (поверхность, соответствующую 1 г адсорбента), чем непористые. По этой причине максимальное количество молекул адсорбата, способных адсорбироваться на 1 г пористого адсорбента (его емкость) много выше чем у непористого. Кроме общего увеличения адсорбции, на пористых адсорбентах происходит конденсация паров адсорбата внутри пор адсорбента при давлениях значительно меньших чем на непористых. Это связано с уменьшением давления насыщенного пара над вогнутыми менисками жидкости внутри капилляров. Это приводит к еще большей, по сравнению с непористыми адсорбентами, степени адсорбции.
1.5. Адсорбция из растворов.
Согласно теории Лэнгмюра, основные закономерности адсорбции из жидких растворов совпадают с закономерностями адсорбции из газов, с той разницей, что поверхность адсорбента всегда целиком занята молекулами растворителя и растворенного вещества. Если обозначить долю поверхности адсорбента, занятую молекулами растворенного вещества как , а молекулами растворителя как 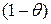 , то скорость адсорбции растворенного вещества, зависящая от числа соударений его молекул с поверхностью адсорбента занятой молекулами растворителя , будет пропорциональна молярной его концентрации. Скорость десорбции будет пропорциональна числу молекул растворенного вещества на единице поверхности адсорбента, то есть . В равновесии скорости адсорбции и десорбции равны, поэтому:
, то скорость адсорбции растворенного вещества, зависящая от числа соударений его молекул с поверхностью адсорбента занятой молекулами растворителя , будет пропорциональна молярной его концентрации. Скорость десорбции будет пропорциональна числу молекул растворенного вещества на единице поверхности адсорбента, то есть . В равновесии скорости адсорбции и десорбции равны, поэтому:
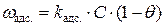 = ,
= ,
откуда:
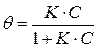 ,
,
где − константа равновесия процесса адсорбции.
Если в растворе находится два растворенных вещества, то отношение степеней их адсорбции зависит как от соотношения их концентраций, так и констант равновесия адсорбции:
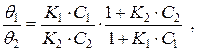
где 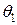 и
и  − степень адсорбции растворенного вещества (1) и (2), соответственно. Чем выше концентрация растворенного вещества и константа равновесия его адсорбции, тем большая доля поверхности занята этим растворенным веществом.
− степень адсорбции растворенного вещества (1) и (2), соответственно. Чем выше концентрация растворенного вещества и константа равновесия его адсорбции, тем большая доля поверхности занята этим растворенным веществом.
Рассмотрим процесс адсорбции растворенного вещества из водного раствора на поверхности активированного угля. Уголь является неполярным адсорбентом, слабо взаимодействующим с молекулами воды. По этой причине поверхностное натяжение на такой поверхности довольно велико. Любое растворенное вещество, менее полярное чем вода, попадая в поверхностный слой уменьшает поверхностное натяжение, так как энергия взаимодействия его с водой или с другой такой же молекулой меньше энергии взаимодействия молекул воды между собой:
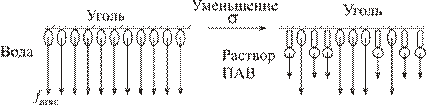
Поскольку при этом уменьшается и энергия Гиббса системы, процесс вытеснения молекул воды с поверхности угля менее полярными молекулами ПАВ, то есть их адсорбция, протекает самопроизвольно. Такие вещества являются поверхностно-активными по отношению к воде. Они накапливается на поверхности угля. Одновременно протекает и их десорбция, степень которой растет с температурой. Величина равновесной адсорбции определяется адсорбционным уравнением Гиббса:
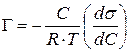 моль/м2,
моль/м2,
где С − молярная концентрация растворенного ПАВ; 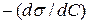 − его поверхностная активность. Для гомологического ряда ПАВ выполняется правило Траубе (см. теоретическое введение). Так, пропионовая кислота СН3СН2СООН адсорбируется при одной и той же концентрации примерно втрое в большей степени чем уксусная кислота СН3СООН, поскольку ее поверхностная активность втрое выше чем у уксусной кислоты. В случае особо мелкопористых адсорбентов правило Траубе не выполняется, поскольку с ростом длины цепочки увеличивается объем молекулы ПАВ и уменьшается число пор, доступных для нее, уменьшается эффективная удельная площадь поверхности адсорбента. В этом случае выполняется так называемое обращенное правило Траубе − чем длиннее цепочка, тем меньше адсорбция.
− его поверхностная активность. Для гомологического ряда ПАВ выполняется правило Траубе (см. теоретическое введение). Так, пропионовая кислота СН3СН2СООН адсорбируется при одной и той же концентрации примерно втрое в большей степени чем уксусная кислота СН3СООН, поскольку ее поверхностная активность втрое выше чем у уксусной кислоты. В случае особо мелкопористых адсорбентов правило Траубе не выполняется, поскольку с ростом длины цепочки увеличивается объем молекулы ПАВ и уменьшается число пор, доступных для нее, уменьшается эффективная удельная площадь поверхности адсорбента. В этом случае выполняется так называемое обращенное правило Траубе − чем длиннее цепочка, тем меньше адсорбция.
Адсорбция карбоновой кислоты на поверхность активированного угля из неполярного растворителя, например бензола, не происходит, поскольку молекулы карбоновой кислоты являются более полярными чем молекулы бензола. Поэтому, попадая в поверхностный слой они увеличивают поверхностное натяжение, то есть энергию системы. Такой процесс самопроизвольно происходить не может. Карбоновая кислота по отношению к бензолу оказывается поверхностно-инактивным веществом и не адсорбируется на поверхности раздела. По этой же причине из водного раствора на поверхности активированного угля не адсорбируются ионы электролитов, взаимодействующие между собой и с молекулами воды с энергией большей, чем молекулы воды между собой.
Как показано ранее, зависимость степени адсорбции от концентрации растворенного вещества описывается уравнением Лэнгмюра:
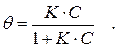
Поскольку 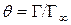 , где
, где 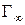 − максимальная адсорбция Гиббса, то:
− максимальная адсорбция Гиббса, то:
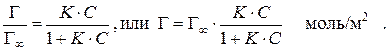
Так как определение удельной площади поверхности адсорбента весьма затруднено, на практике часто пользуются величиной адсорбции не на единице поверхности адсорбента, а на единице его массы. В этом случае степень адсорбции выражают в молях адсорбата на единицу массы адсорбента (х/m) молей/г и уравнение Лэнгмюра приобретает вид:
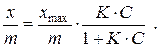
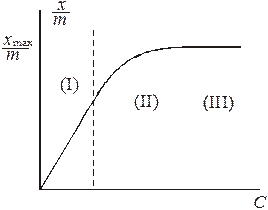 График зависимости степени адсорбции от концентрации растворенного ПАВ может быть условно разбит на три части. При малых концентрациях ПАВ, то есть при
График зависимости степени адсорбции от концентрации растворенного ПАВ может быть условно разбит на три части. При малых концентрациях ПАВ, то есть при 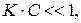 зависимость является линейной (I). В этой области концентраций ПАВ поверхность адсорбента занята в основном молекулами растворителя. И наоборот, при достаточно высокой концентрации ПАВ, при
зависимость является линейной (I). В этой области концентраций ПАВ поверхность адсорбента занята в основном молекулами растворителя. И наоборот, при достаточно высокой концентрации ПАВ, при 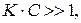 вся поверхность адсорбента занята молекулами ПАВ, степень адсорбции равна максимальной и не зависит от концентрации ПАВ (III). В промежуточной области, при
вся поверхность адсорбента занята молекулами ПАВ, степень адсорбции равна максимальной и не зависит от концентрации ПАВ (III). В промежуточной области, при 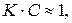 зависимость величины адсорбции от концентрации ПАВ является нелинейной (II).
зависимость величины адсорбции от концентрации ПАВ является нелинейной (II).
Как и в случае адсорбции газов, увеличение температуры приводит к росту скорости десорбции и соответствующему уменьшению степени адсорбции при данной равновесной концентрации ПАВ. Величина максимальной адсорбции определяется величиной удельной поверхности адсорбента и размерами молекул адсорбата, поэтому не зависит от температуры.
Нелинейный участок изотермы адсорбции описывается эмпирическим уравнением Фрейндлиха:
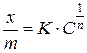 ,
,
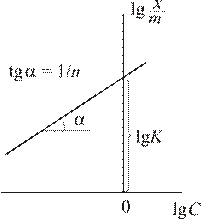 где величина константы К зависит от условий адсорбции, свойств адсорбента (от его удельной поверхности), размеров молекул адсорбата, а величина константы 1/ n составляет 0.3–0.5. Для конкретных условий адсорбции константы уравнения Фрейндлиха легко могут быть найдены путем измерения степени адсорбции при нескольких равновесных концентрациях ПАВ. Логарифмирование уравнения Фрейндлиха приводит к уравнению прямой в координатах
где величина константы К зависит от условий адсорбции, свойств адсорбента (от его удельной поверхности), размеров молекул адсорбата, а величина константы 1/ n составляет 0.3–0.5. Для конкретных условий адсорбции константы уравнения Фрейндлиха легко могут быть найдены путем измерения степени адсорбции при нескольких равновесных концентрациях ПАВ. Логарифмирование уравнения Фрейндлиха приводит к уравнению прямой в координатах 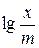 −
− 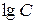 :
:
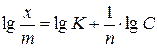 .
.
Построив соответствующий график, по величине отсекаемого отрезка, равного 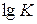 , можно найти константу К, а тангенс угла наклона графика дает величину 1/ n.
, можно найти константу К, а тангенс угла наклона графика дает величину 1/ n.
1.6. Ход работы и обработка полученных данных.
Приготовление исходных растворов уксусной кислоты.
1.6.1. В пять нумерованных колб с помощью мерной колбы налить по 100 мл дистиллированной воды.
1.6.2. С помощью мерной колбы налить в колбу № 1 100 мл исходного (по заданию) раствора с концентрацией уксусной кислоты 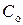 , сполоснуть мерную колбу образовавшимся раствором с концентрацией уксусной кислоты равной
, сполоснуть мерную колбу образовавшимся раствором с концентрацией уксусной кислоты равной 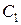 , вылить его обратно в колбу № 1.
, вылить его обратно в колбу № 1.
1.6.3. С помощью мерной колбы 100 мл раствора из колбы № 1 вылить в колбу № 2, ополоснуть мерную колбу образовавшимся раствором, вылить обратно в колбу № 2.
1.6.4. Аналогично 100 мл раствора из колбы № 2 с помощью мерной колбы перелить в колбу № 3 и т.д. Из колбы № 5 лишние 100 мл раствора вылить в раковину.
Проведение адсорбции уксусной кислоты на активированном угле.
1.6.5. На технических весах взять пять навесок по 2.00±0.01 г активированного угля (m), всыпать их в колбы № 1 − № 5, записать время.
1.6.6. В течение ~30 минут поочередно встряхивать колбы для ускорения установления равновесия адсорбции.
1.6.7. По истечении ~30 минут профильтровать растворы из колб № 1 − № 5 через складчатые фильтры в соответствующие пронумерованные колбы, отбросив в каждом случае небольшую порцию фильтрата.
Определение исходных и равновесных концентраций растворов уксусной кислоты.
1.6.8. Путем потенциометрического титрования раствором щелочи известной концентрации (см. инструкцию на рабочем месте) определить равновесную концентрацию уксусной кислоты в каждом из пяти фильтратов (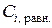 ). Для каждого фильтрата провести три параллельных титрования, взять среднее арифметическое, заполнить табл. 1.
). Для каждого фильтрата провести три параллельных титрования, взять среднее арифметическое, заполнить табл. 1.
1.6.9. Аналогично определить концентрацию исходного раствора уксусной кислоты (), вычислить исходные концентрации растворов в колбах № 1 − № 5: 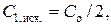
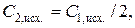
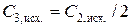 и т.д., внести полученные данные в табл. 2.
и т.д., внести полученные данные в табл. 2.
Определение констант уравнения Фрейндлиха.
1.6.10. Вычислить исходные 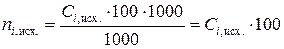 и равновесные
и равновесные 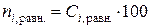 , ммоль, содержания уксусной кислоты в 100 мл раствора, а также равновесные величины адсорбции
, ммоль, содержания уксусной кислоты в 100 мл раствора, а также равновесные величины адсорбции 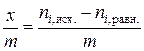 , ммоль/г, заполнить табл. 2.
, ммоль/г, заполнить табл. 2.
1.6.11. Построить графики зависимости степени адсорбции от равновесной концентрации уксусной кислоты в координатах 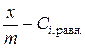 и
и 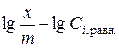 .
.
1.6.12. Графически определить коэффициенты уравнения Фрейндлиха − К и 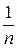 .
.
Таблица 1.
| № раствора | Объем раствора на титрование, мл | Эквивалентный объем титранта (NaOH), мл | Норм. титранта, г-экв/л | 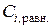 , г-экв/л , г-экв/л | ||
| Средн. | ||||||
| …. |
Таблица 2.
| № раствора | Конц. уксусной кислоты, г-экв./л | Содерж. уксусной к-ты в 100 мл. раствора, ммоль | 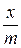 , ммоль/г , ммоль/г | lg Ci, равн. | lg (x/m) . | ||
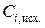 | | 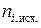 | 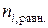 | ||||
| …. |
Контрольные вопросы
1. Виды межмолекулярного взаимодействия. Энергия притяжения и отталкивания, потенциальная кривая взаимодействия двух частиц, зависимость энергии взаимодействия от структуры частиц.
2. Поверхностное натяжение на границе раздела фаз. Причина возникновения, влияние различных факторов на величину поверхностного натяжения.
3. Мономолекулярная и полимолекулярная адсорбция на твердой поверхности. ПАВ и ПИАВ, влияние различных факторов на степень адсорбции. Уравнение Лэнгмюра, уравнение Фрейндлиха.
4. Нахождение констант уравнения Фрейндлиха для адсорбции карбоновой кислоты из водного раствора на поверхности активированного угля.
Литература
1. Герасимов Я.И., Древиг В.П., Еремин Е.Н. и др. Курс физической химии. Том I. –М: ГХИ, 1963. С. 435 – 455, 513 – 542.
2. Григоров О.Н. и др. Руководство к практическим работам по коллоидной химии. –М., –Л.: Химия, 1964. С. 86 – 92, 107 – 120.
3. Воюцкий С.С. Курс коллоидной химии. –М.: Химия, 1976. С. 81 – 99, 137 – 145.
РАБОТА № 8
ИССЛЕДОВАНИЕ ЗАВИСИМОСТИ ПОВЕРХНОСТНОГО НАТЯЖЕНИЯ РАСТВОРОВ ПАВ И АДСОРБЦИИ ИХ НА ГРАНИЦЕ РАЗДЕЛА ЖИДКОСТЬ – ГАЗ ОТ КОНЦЕНТРАЦИИ ПАВ
1. Теоретическая часть
1.1. Адсорбция растворенных веществ на поверхности раздела жидкость – газ. Изотерма адсорбции, зависимость поверхностного натяжения раствора от концентрации ПИВ и ПИАВ.
При постоянной температуре и постоянном давлении энергия гетерогенной системы из двух фаз, разделенных поверхностью раздела, складывается из энергии Гиббса фаз (I) и (II), а также избыточной энергии Гиббса поверхности раздела:
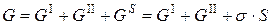
Соответственно, изменение энергии такой системы также является суммой:
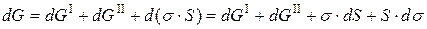 .
.
Самопроизвольными являются все процессы, приводящие к уменьшению энергии Гиббса системы (dG < 0), При постоянстве энергий Гиббса фаз (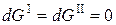 ) такими процессами являются процессы, ведущие либо к уменьшению площади поверхности раздела (
) такими процессами являются процессы, ведущие либо к уменьшению площади поверхности раздела (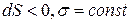 , так капля ртути самопроизвольно принимает форму шара, имеющего наименьшую поверхность при данном объеме), либо к уменьшению поверхностного натяжения при постоянной величине поверхности раздела (
, так капля ртути самопроизвольно принимает форму шара, имеющего наименьшую поверхность при данном объеме), либо к уменьшению поверхностного натяжения при постоянной величине поверхности раздела (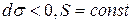 ). Поскольку наличие ПАВ в поверхностном слое приводит к уменьшению поверхностного натяжения, процесс увеличения его концентрации на поверхности раздела по сравнению с концентрацией в объеме является термодинамически выгодным и самопроизвольным.
). Поскольку наличие ПАВ в поверхностном слое приводит к уменьшению поверхностного натяжения, процесс увеличения его концентрации на поверхности раздела по сравнению с концентрацией в объеме является термодинамически выгодным и самопроизвольным.
Процесс концентрирования, или сгущения растворенного вещества на поверхности раздела фаз называется адсорбцией. Этот процесс сопровождается уменьшением энергии системы за счет уменьшения избыточной энергии поверхности раздела фаз. При отсутствии теплового движения и, соответственно, диффузии, растворенное вещество заполняло бы всю поверхность раздела фаз. Процесс адсорбции, однако, всегда сопровождается процессом десорбции, обусловленным диффузией ПАВ с поверхности раздела в объем фазы. При данной концентрации ПАВ и температуре устанавливается равновесие между адсорбцией и десорбцией. Рост концентрации ПАВ в растворе увеличивает скорость адсорбции, увеличение температуры системы увеличивает скорость десорбции. Соответственно этому, с ростом концентрации ПАВ в растворе степень его адсорбции на поверхности растет, а с ростом температуры – уменьшается.
Величину степени адсорбции легко вычислить по уравнению Гиббса, из которого следует, что с ростом концентрации ПАВ и его поверхностной активности степень адсорбции увеличивается, а с ростом температуры – уменьшается:
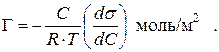
Аналогичная зависимость степени адсорбции ПАВ от его концентрации выражается уравнением изотермы адсорбции Лэнгмюра:
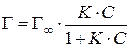 ,
,
где Г∞ – максимальное количество молей ПАВ, способное адсорбироваться на единице площади поверхности раздела; К – константа равновесия адсорбции, зависящая от теплоты адсорбции и, соответственно, от поверхностной активности ПАВ и температуры. Величина Г∞ не зависит от температуры и определяется лишь эффективным сечением молекулы ПАВ (чем больше сечение, тем меньше молекул может поместиться на единице площади поверхности раздела, тем меньше Г∞).
Графики изотерм адсорбции имеют следующий вид:
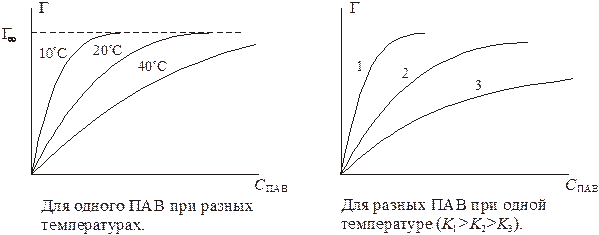
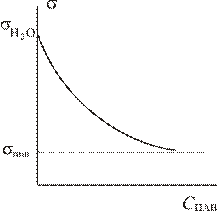 В соответствии с ростом содержания молекул ПАВ в поверхностном слое, то есть с увеличением Г, поверхностное натяжение на границе раздела фаз уменьшается. При максимальной адсорбции Г∞ , на поверхности раздела образуется мономолекулярный слой молекул ПАВ, причем полярными, гидрофильными группами они обращены к жидкой фазе, а гидрофобными – к газовой фазе. Величина поверхностного натяжения принимает минимальное значение
В соответствии с ростом содержания молекул ПАВ в поверхностном слое, то есть с увеличением Г, поверхностное натяжение на границе раздела фаз уменьшается. При максимальной адсорбции Г∞ , на поверхности раздела образуется мономолекулярный слой молекул ПАВ, причем полярными, гидрофильными группами они обращены к жидкой фазе, а гидрофобными – к газовой фазе. Величина поверхностного натяжения принимает минимальное значение 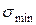 , соответствующее поверхностному натяжению мономолекулярного слоя ПАВ. Математически зависимость σ от концентрации ПАВ выражается уравнением Шишковского:
, соответствующее поверхностному натяжению мономолекулярного слоя ПАВ. Математически зависимость σ от концентрации ПАВ выражается уравнением Шишковского:
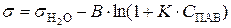 ,
,
где В – коэффициент, определяемый геометрическими размерами молекулы ПАВ и не зависящий от его поверхностной активности; К – константа равновесия адсорбции.
Величина максимальной адсорбции Г∞ легко находится графическим способом из уравнения изотермы адсорбции. Действительно, из уравнения Лэнгмюра следует, что
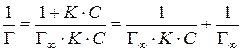 .
.
Если умножить обе части этого равенства на концентрацию ПАВ оказывается, что зависимость С/ Г от С является линейной:
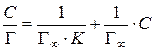 .
.
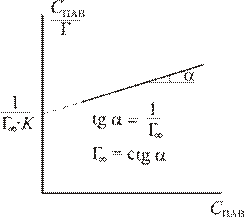 Используя экспериментальные данные строят график в координатах С ПАВ / Г – С ПАВ, из наклона которого определяют Г∞, а из отсекаемого отрезка – величину константы равновесия адсорбции.
Используя экспериментальные данные строят график в координатах С ПАВ / Г – С ПАВ, из наклона которого определяют Г∞, а из отсекаемого отрезка – величину константы равновесия адсорбции.
Поскольку при предельной адсорбции Г∞ число молей ПАВ в адсорбционном слое, соответствующем 1 м2 поверхности раздела много больше соответствующей величины в том же объеме внутри фазы (n S>> n), то Г∞ = n S – n = n S. Следовательно, число молекул ПАВ на единице площади поверхности раздела фаз при максимальной адсорбции составляет n S· N A = Г∞· N A, где N A = 6.02·1023 1/моль – число Авогадро. Площадь сечения одной молекулы ПАВ, определяемая размером полярной группы, погруженной в жидкую фазу, составляет:
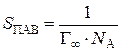 , м2.
, м2.
Объем адсорбционного слоя, соответствующий 1 м2 поверхности раздела можно вычислить как произведение площади на толщину слоя, равную эффективной длине молекулы ПАВ. Его масса равна произведению объема на плотность ПАВ: 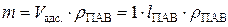 . В то же время масса поверхностного слоя ПАВ на 1 м2 поверхности раздела равна произведению массы одного моля ПАВ (ММПАВ) на число молей ПАВ на 1 м2 поверхности раздела при предельной адсорбции (n S = Г∞):
. В то же время масса поверхностного слоя ПАВ на 1 м2 поверхности раздела равна произведению массы одного моля ПАВ (ММПАВ) на число молей ПАВ на 1 м2 поверхности раздела при предельной адсорбции (n S = Г∞): 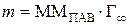 . Следовательно:
. Следовательно:
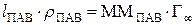 , откуда
, откуда 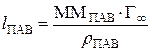 , м.
, м.
Таким образом, определив предельную адсорбцию можно вычислить геометрические параметры молекулы ПАВ – ее длину и площадь поперечного сечения.
В то время как ПАВ уменьшают поверхностное натяжение на поверхности раздела фаз, ПИАВ либо не влияют на величину поверхностного натяжения, либо слабо ее повышают. Зависимость величины 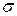 от концентрации ПАВ в растворе является линейной:
от концентрации ПАВ в растворе является линейной:
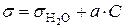 .
.
Величина поверхностной активности ПИАВ является отрицательной. Отрицательной является и величина адсорбции. Графики зависимости и Г от концентрации ПИАВ имеют вид:
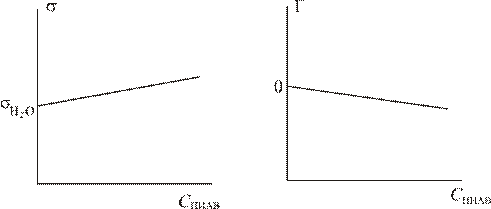
1.3. Измерение поверхностного натяжения на границе раздела "жидкость – газ".
1.3.1. Метод капиллярного поднятия.
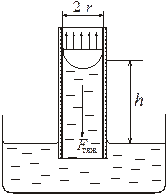 При идеальном смачивании материала капилляра жидкосью, сила, втягивающая столб жидкости в капилляр, равна произведению поверхностного натяжения (сила, приходящаяся на единицу длины контура поверхности раздела) на длину окружности мениска жидкости:
При идеальном смачивании материала капилляра жидкосью, сила, втягивающая столб жидкости в капилляр, равна произведению поверхностного натяжения (сила, приходящаяся на единицу длины контура поверхности раздела) на длину окружности мениска жидкости:
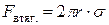 .
.
В равновесии эта сила уравновешена силой тяжести столба жидкости:
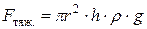 .
.
Таким образом: 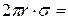
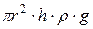 , откуда:
, откуда:
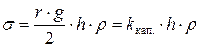 ,
,
где 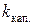 – константа капилляра, h – высота поднятия, а ρ – плотность жидкости. Константу капилляра легко найти, измерив высоту поднятия и плотность жидкости с точно известной величиной поверхностного натяжения (чаще всего дистиллированной воды):
– константа капилляра, h – высота поднятия, а ρ – плотность жидкости. Константу капилляра легко найти, измерив высоту поднятия и плотность жидкости с точно известной величиной поверхностного натяжения (чаще всего дистиллированной воды):
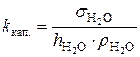 .
.
1.3.2. Метод счета капель.
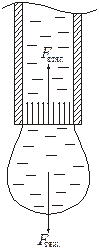 В этом методе применяют несложный прибор – сталагмометр, представляющий собой пипетку с двумя рисками. Объем жидкости между рисками точно известен (
В этом методе применяют несложный прибор – сталагмометр, представляющий собой пипетку с двумя рисками. Объем жидкости между рисками точно известен (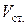 ). Из сталагмометра отдельными каплями вытекает исследуемая жидкость. Отрыв каждой капли от капилляра, находящегося на конце сталагмометра, происходит при равенстве силы ее тяжести (F тяж.)и силы, втягивающей ее в капилляр (F втяг.):
). Из сталагмометра отдельными каплями вытекает исследуемая жидкость. Отрыв каждой капли от капилляра, находящегося на конце сталагмометра, происходит при равенстве силы ее тяжести (F тяж.)и силы, втягивающей ее в капилляр (F втяг.):
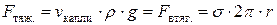 .
.
Объем одной капли (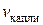 ) определяют подсчитав число капель N, соответствующее объему сталагмометра:
) определяют подсчитав число капель N, соответствующее объему сталагмометра: 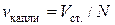 . Тогда:
. Тогда:
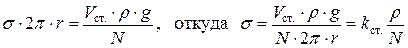 ,
,
где 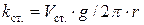 – константа сталагмометра, которую легко найти, подсчитав число капель дистиллированной воды, соответствующее объему сталагмометра:
– константа сталагмометра, которую легко найти, подсчитав число капель дистиллированной воды, соответствующее объему сталагмометра: 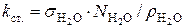 .
.
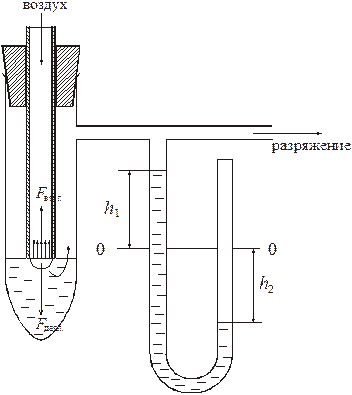
1.3.3. Метод максимального давления в пузырьке (метод Ребиндера).
В этом методе пузырек воздуха продавливают через капилляр, касающийся поверхности раздела с исследуемой жидкостью в приборе Ребиндера. Необходимое для этого разряжение оказывается пропорциональным поверхностному натяжению на границе раздела фаз, поскольку продавливание пузырька происходит при равенстве сил втягивания пузырька внутрь капилляра (F втяг.) и выталкивания его за счет сил давления (F давл.):
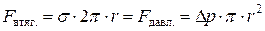 .
.
Отсюда: 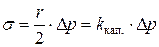 .
.
Константу капилляра k кап. 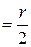 легко найти, измерив
легко найти, измерив 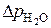 = h 1 + h 2, продавливая пузырьки воздуха через дистиллированную воду, поверхностное натяжение для которой (
= h 1 + h 2, продавливая пузырьки воздуха через дистиллированную воду, поверхностное натяжение для которой (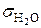 ) точно измерено:
) точно измерено:
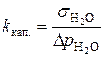 .
.

