 2015-07-21
2015-07-21 609
609В рамках настоящей работы создана экспериментальная установка (рис. 1), позволяющая реализовать сверхкритический флюидный экстракционный (СКФЭ) процесс с целью регенерации катализатора оксид алюминия активный. Данная экспериментальная установка, имеющая возможность использования как чистого, так и модифицированного флюидного экстрагента, защищена патентом на полезную модель РФ [7].
Процесс модификации экстрагента может осуществляться добавлением сорастворителя в диоксид углерода перед подачей его в ячейку.
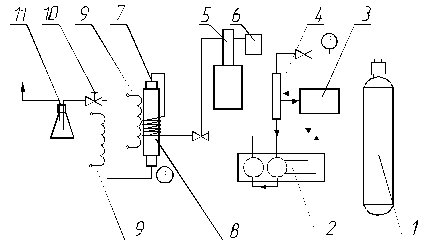
Рис. 1 Схема экспериментальной установки
1 – баллон с СО2, 2 – жидкостной насос для подачи СО2, 3 – холодильный агрегат, 4 – конденсатор, 5 – шприцевой насос для подачи сорастворителя; 6 – емкость для сорастворителя; 7 – экстракционная ячейка; 8 – теплообменник; 9 – ТЭНы, 10 – регулирующий вентиль (с функциями дросселирующего устройства), 11 – сборник экстракта
Установка смонтирована на базе жидкостного плунжерного насоса марки «LIQUPUMP 312/1». Для нормальной работы насоса необходимо обеспечивать подачу жидкого диоксида углерода на контур всасывания. Сжижение газа происходит в конденсаторе и непосредственно в насосе за счет циркуляции хладагента в рубашке охлаждения насоса и межтрубном пространстве конденсатора. Конструктивной особенностью насоса является наличие двух насосных головок, плунжеры которых, работая последовательно, сглаживают пульсацию потока флюида. Благодаря этому достигается высокая степень равномерности подачи СКФ.
Шприцевой насос высокого давления марки СФЭ-400 служит для подачи модификатора из емкости. Необходимая концентрация модификатора в сверхкритическом (СК) СО2 (1 ÷ 9%) устанавливается путем регулирования расхода сорастворителя в диапазоне 0,01 ÷ 11 мл/мин.
Обратные клапаны, используемые в системе, предотвращают возврат газа и жидкости обратно в насосы.
Экстракционная ячейка помещена в теплообменник, представляющий собой толстостенную медную трубку. Нагрев осуществляется за счет ТЭНа, намотанного на теплообменник. Температура экстракционной ячейки поддерживается с помощью электронного измерительного регулятора 2ТРМ1. Для контроля температуры используются хромель-копелевые термопары, установленные на концах ячейки. Точность измерения температуры оценивается в пределах ±0,05 °С.
Для определения изменения массы катализатора, а соответственно и количества извлеченных дезактивирующих веществ, производится его взвешивание до и после эксперимента на электронных весах «САРТО ВЛТ-150-П» с точностью измерений ± 10–6 кг.
Состав экстракта отработанного катализатора определяли методом газожидкостной хроматографии с использованием хроматографа «Хроматэк-Кристалл-2000» с пламенно-ионизационным детектором на капиллярной колонке CP-WAX 52 длиной 50 м и диаметром 0,32 мм при температуре 90 °С. В качестве газа-носителя использовался азот. Жидкая проба вводилась в испаритель в количестве 0,6 мкл; температура испарителя варьировалась в пределах 10 – 200 °С; температура детектора составляла 260 °С. Анализ хроматограммы проводился использованием программного комплекса NetChom v1.5.
Образцы дезактивированного катализатора исследованы методами термогравиметрии, деривативной термогравиметрии (ТГ – ДТГ) в комплексной постановке. Исследования проведены на дериватографе системы «F.PAULIK» интервале температур 20 – 850 °С в открытых тарельчатых платиновых тиглях в воздушной среде из навесок в 600 мг. Абсолютная погрешность определения температуры ±5 °С, а относительная погрешность измерения массы при заданной чувствительности 0,2 мг составляет ±0,5 %.
Для проведения ИК-спектроскопических исследований загрязняющих отложений образец дезактивированного катализатора заливают хлороформом. Через 2 часа полученный экстракт переносят на пластинку из NaCl, после чего выпаривают хлороформ и получают тонкую пленку исследуемого вещества. Пластинку помещают в ИК-Фурье спектрофотометр Vector 33 и выписывают спектры в диапазоне длин волн 600 ÷ 4000 см–1 при комнатной температуре в течение 1 мин.
Кроме того, в рамках данного исследования проведены измерения селективности, то есть способности протекания химической реакции в определённом направлении, а именно свойств получать те продукты реакции, на которые направлена химическая реакция и конверсии регенерированного катализатора на установке, схема которой представлена на рис. 2.
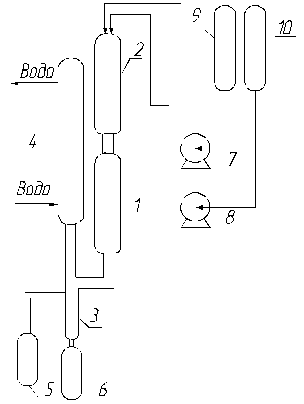
Рис. 2 Установка для исследования процессов парофазной дегидратации МФК
1 – реактор, 2 – испаритель, 3 – сепаратор, 4 – обратный холодильник, 5 – приемник углеводородной фракции дистиллята (катализата), 6 – приемник водной фракции дистиллята, 7 – дозирующий насос подачи МФК, 8 – дозирующий насос подачи воды, 9 – емкость подачи МФК, 10 – емкость подачи воды
Из емкости 9 насосом 7 в испаритель 2, заполненный битым кварцевым стеклом, подается фракция МФК. Туда же подается дистиллированная вода из емкости 10 насосом 8 и происходит их испарение при температуре 300 °С. Пары воды и МФК из испарителя поступают в реактор 1, заполненный катализатором, где протекает реакция гидратации. Из нижней части реактора продукты реакции поступают в обратный холодильник 4, где происходит их конденсация. Далее конденсат стекает в сепаратор 3, где разделяется на водную и углеводородную фракции. Из сепаратора углеводородная фаза (верхний слой) стекает в приемник 5, а водная фаза (нижний слой) в приемник 6. Затем катализат из емкости 5 исследуется хроматографическим анализом. Условия проведения экспериментов: весовое соотношение МФК:Н2О = 1:1, подача МФК и Н2О – 45 см3/ч, объем загруженного в реактор катализатора – 100 см3. Длительность процесса парофазной дегидратации МФК составляет 10 часов.








