2015-10-16
2015-10-16 621
621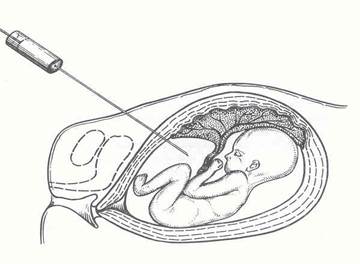
Рис. 4 – Амніоцентез
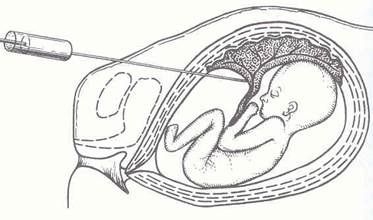
Рис. 5 - Кордоцентез
Отриманий при інвазивній ПД матеріал досліджують за допомогою лабораторних методів (цитогенетичних, молекулярно-генетичних, біохімічних, імуноцитохімічних).
При застосуванні непрямих методів ПД стан плода оцінюють по біохімічним показникам у крові та сечі вагітної, даним акушерсько-гінекологічного анамнезу. Бажаним, а для жінок високого ризику обов’язковим, є медико-генетичне консультування, яке доповнене результатами мікробіологічного, імунологічного, ендокринного обстеження.
Пренатальне дослідження здійснюється у відповідності з трьома етапами:
1 етап включає комбінований тест в 10 тижнів вагітності – ранній пренатальний скринінг (визначення рівня РАРР і ХГЧ) для виявлення хромосомних хвороб і вад нервової трубки; ультразвукове дослідження в 10 тижнів вагітності з оцінкою комірцевого простору, носових кісток та швидкості кровоплину у венозній протоці, що є високоінформативними ознаками, пошук плодових еритробластів із крові вагітних жінок та їх ДНК-аналіз.
2 етап – потрійний тест в терміні 16-20 тижнів вагітності включає дослідження крові на альфафетопротеїн, хоріонічний гонадотропін і незв’язаний естріол. В 18-24 тижні ультразвукове дослідження з оцінкою морфогенетичної структури всіх органів плоду. Термін вагітності для подальшого ультразвукового слідкування за розвитком плоду визначається індивідуально. Амніоцентез проводиться в 14-21 тиждень, кордоцентез – починаючи з 21 тижня.
3 етап – ультразвукове дослідження в 32-36 тижнів з обов’язковою доплерографією системи “плід-плацента-мати”.
Отримані під час інвазивних процедур біологічних зразків плода підлягають дослідженню за допомогою спеціальних лабораторних методів. Серед них, перш за все, цитогенетичне дослідження – вивчення хромосомного набору (каріотипу) клітин отриманого зразка на всіх етапах ембріогенезу. В залежності від терміну вагітності досліджуються клітини хоріону, плаценти, лімфоцити пуповиної крові, всі вони мають плодове походження і відповідають його генетичним характеристикам.
Молекулярні методи використовуються для діагностики генних хвороб. Вони поділяються на прямі і непрямі в залежності від того, що досліджується мутантний ген чи поліморфні сайти рестрикції, так звані молекулярні маркери. При проведенні молекулярної пренатальної діагностики необхідно:
· точний клінічний діагноз;
· адекватно розрахований ризик захворювання плода;
· визначення оптимального терміну ПД;
· забезпечення можливості отримати біологічні зразки плода;
· розробка чітких рекомендацій після проведення ПД;
· наявність скринуючих програм ДНК-діагностики.
За допомогою вказаних методів клініко-генетичної і молекулярної діагностики можна пренатально встановити діагнози таких моногенних хвороб (табл.4):
Таблиця 4.
| № п/п | Спадкові захворювання |
| 1. | Агамаглобулінемія |
| 2. | Адреногенітальний синдром |
| 3. | Альбінізм тип ОСА 1 |
| 4. | Альпорта синдром |
| 5. | Альфа-1-Антитрипсину недостатність |
| 6. | Аміотрофія спінальна Верднига-Гоффмана, Кугельберга-Веландера |
| 7. | Аміотрофія невральна Шарко-Марі-Тута |
| 8. | Аміотрофія спінальна Х-зчеплена |
| 9. | Анжельмана синдром |
| 10. | Апера синдром |
| 11. | Атаксія Фридрейха |
| 12. | Ахондроплазія |
| 13. | Беквіта-Відемана синдром |
| 14. | Бета-таласемія |
| Віллебранда хвороба | |
| 16. | Вроджена контрактурна арахнодактілія |
| 17. | Вроджена м'язова дистрофія, тип Фукуяма |
| 18. | Гемофілія А |
| 19. | Гемофілія В |
| 20. | Гепатолентикулярна дегенерація (хвороба Вільсона-Коновалова) |
| 21. | Гіперхолестерінемія сімейна |
| 22. | Гіпофизарний нанізм (дефіцит гормону росту) |
| 23. | Глікогенози |
| 24. | Глухота нейросенсорна несиндромальна |
| 25. | Грейга синдром |
| 26. | Дефіцит ацил-КоА дегідрогенази |
| 27. | Довгого QT синдром |
| 28. | Жильбера синдром |
| 29. | Зонулярна катаркта |
| 30. | Коффіна-Лоурі синдром |
| 31. | Криглера-Найара синдром |
| 32. | Лімфопроліферативний синдром, Х-зчеплений (хвороба Дункана, синдром Пуртільо) |
| 33. | Лімфидема Мінроя |
| 34. | Луі-Барр синдром (телеангіоектазія) |
| 35. | Леш-Ніхана синдром |
| 36. | Мартіна-Белл (ламкої Х-хромосоми) синдром |
| 37. | Марфана синдром |
| 38. | Міллера-Дікера синдром |
| 39. | Мілроя синдром |
| 40. | Міодистрофія Дюшенна/Беккера |
| 41. | Міодистрофія Емері-Дрейфуса |
| 42. | Міотонічна дистрофія |
| 43. | Муковісцидоз |
| 44. | Мукополісахарідози |
| 45. | Несиндромальна нейросенсорна приглухуватість |
| 46. | Ніймегена синдром |
| 47. | Норрі хвороба |
| 48. | Окулофарінгеальна міодистрофія |
| 49. | Періодична хвороба |
| 50. | Псевдоахондропластична дисплазія |
| 51. | Прадера-Віллі синдром |
| 52. | Рецесивний полікістоз нирок |
| 53. | Ретінобластома |
| 54. | Сміта-Лемлі-Опітца синдром |
| 55. | Спастична параплегія Штрюмпеля |
| 56. | Спино-Бульбарна м'язова атрофія (хвороба Кеннеді) |
| 57. | Тестикулярної фемінізації синдром |
| 58. | Унферріхта-Лундберга хвороба |
| 59. | Фенілкетонурія |
| 60. | Хантера хвороба |
| 61. | Хольт-Орана синдром |
| 62. | Хорея Гентингтона |
| 63. | Ектодермальна ангідротична дисплазія |
| 64. | Елерса-Данло (класичний тип) синдром |