2015-10-13
2015-10-13 2034
2034ность (ХЛЖН) - это медленно формирующееся патологическое состояние, при котором нагрузка на левый желудочек превышает его способность совершать работу. Следует отметить, что этиологические факторы острой и хронической недостаточности сердца существенно различаются. ХЛЖН осложняет течение только хронических заболеваний сердца и сосудов.
Правожелудочковая недостаточность характеризуется развитием застойных явлений в большом круге кровообращения. При этом увеличивается кровенаполнение печени и, соответственно, ее размеры, нарушается экскреторная функция почек, происходит задержка воды в организме и появляются периферические отеки. Различают острую и хроническую правожелудоч-ковую недостаточность. Наиболее частой причиной острой правожелудочковой недостаточности является распространение крупноочагового инфаркта левого желудочка на правые отделы сердца, реже - изолированный некроз миокарда правого желудочка. Очень часто причиной правожелудочковой недостаточности является легочная гипертензия, которая также может развиваться остро и хронически (разд. 14.2.3).
К весьма распространенным проявлениям перегрузки правого желудочка относятся легочное сердце и эмболия легочной артерии.
Понятие «легочное сердце» включает в себя легочную гипертензию, гипертрофию правого желудочка, его дилатацию и сердечную недостаточность. Однако в ряде случаев легочное сердце может проявляться только некоторыми из вышеназванных признаков. Так, длительное повышение АД в легочных сосудах обычно приводит к гипертрофии правого желудочка с последующим развитием сердечной недостаточности по большому кругу. Если легочная гипертензия быстро прогрессирует, то гипертрофия правого желудочка не успевает развиться и дилата-ция непосредственно переходит в правожелудоч-ковую недостаточность. Такая ситуация может иметь место и при тромбоэмболии легочной артерии, пневмотораксе, астматическом статусе и распространенной пневмонии, когда АД в малом круге повышается в течение нескольких суток или даже часов.
Синдром эмболии легочной артерии - это патологическое состояние, которое развивается при попадании эмболов в русло легочной артерии и характеризуется болями в грудной
Механизмы компенсации гемодинамики при сердечной недостаточности
Здоровый организм обладает многообразными механизмами, обеспечивающими своевременную разгрузку сосудистого русла от избытка кидкости. К этим механизмам относятся: активация выделительной функции почек, депонирование крови в печени и селезенке, потоотде-тение, испарение воды со стенок легочных альвеол, компенсаторное изменение артериального давления, насосной функции сердца и др. В условиях сердечной недостаточности «включаются» компенсаторные механизмы, направленные на сохранение нормальной гемодинамики. Эти механизмы в условиях острой и хронической недостаточности кровообращения имеют много эбщего, вместе с тем между ними отмечаются существенные различия.
Механизмы компенсации гемодинамических нарушений при острой сердечной недостаточности
На начальной стадии систолической дисфункции желудочков сердца включаются интракар-циальные факторы компенсации сердечной недостаточности, важнейшим из которых является механизм Франка - Старлинга. Реализацию эго можно представить следующим образом. Нарушение сократительной функции сердца вле- ier за собой уменьшение ударного объема крови \ гипоперфузию почек. Это способствует акти-зации ренин-ангиотензин-альдостероновой сис-гемы, вызывающей задержку воды в организме я увеличение объема циркулирующей крови. В условиях возникшей гиперволемии происходит усиленный приток венозной крови к сердцу, увеличение диастолического кровенаполнения желудочков, растяжение миофибрилл миокарда а компенсаторное повышение силы сокращения сердечной мышцы, которое обеспечивает прирост ударного объема. Однако если конечное диасто-гаческое давление повышается более чем на 18- 12 мм рт. ст., возникает чрезмерное перерастя-кение миофибрилл. В этом случае компенсаторный механизм Франка - Старлинга перестает действовать, а дальнейшее увеличение конечно-'О диастолического объема или давления вызывает уже не подъем, а снижение УО.
Наряду с внутрисердечными механизмами
компенсации при острой левожелудочковой недостаточности запускаются разгрузочные экст-ракардиальные рефлексы, способствующие возникновению тахикардии и увеличению минутного объема крови (МОК). Одним из наиболее важных сердечно-сосудистых рефлексов, обеспечивающих увеличение МОК, является рефлекс Бейибриджа - увеличение частоты сердечных сокращений в ответ на увеличение объема циркулирующей крови. Этот рефлекс реализуется при раздражении механорецепторов, локализованных в устье полых и легочных вен. Данные механорецепторы являются афферентными окончаниями вагуса, и их раздражение передается на центральные симпатические ядра продолговатого мозга, в результате чего происходит повышение тонической активности симпатического звена вегетативной нервной системы и развивается рефлекторная тахикардия. Рефлекс Бейибриджа направлен на увеличение минутного объема крови.
Рефлекс Бецольда - Яриша - это рефлекторное расширение артериол большого круга кровообращения в ответ на разражение меха-но- и хеморецепторов, локализованных в желудочках и предсердиях. В результате возникает гипотония, которая сопровождается брадикар-дией и временной остановкой дыхания. В реализации этого рефлекса принимают участие афферентные и эфферентные волокна п. vagus. Этот рефлекс направлен на разгрузку левого желудочка.
К числу компенсаторных механизмов при острой сердечной недостаточности относится и повышение активности симпатоадреналовой системы, одним из звеньев которого является высвобождение норадреналина из окончаний симпатических нервов, иннервирующих сердце и почки. Наблюдаемое при этом возбуждение (3-адренорецепторов миокарда ведет к развитию тахикардии, а стимуляция подобных рецепторов в клетках юкстагломерулярного аппарата вызывает усиленную секрецию ренина. Другим стимулом секреции ренина является снижение почечного кровотока в результате вызванной катехоламинами констрикции артериол почечных клубочков. Компенсаторное по своей природе усиление адренергического влияния на миокард в условиях острой сердечной недостаточности направлено на увеличение ударного и минутного объемов крови. Положительный
инотропный эффект оказывает также ангиотен-зин-Н. Однако эти компенсаторные механизмы могут усугубить сердечную недостаточность, если повышенная активность адренергической и ре-нин-ангиотензиновой системы сохраняется достаточно продолжительное время (более 24 ч).
Все сказанное о механизмах компенсации сердечной деятельности в одинаковой степени относится как к лево-, так и к правожелудочковой недостаточности. Исключением является рефлекс Ларина, действие которого реализуется только при перегрузке правого желудочка, наблюдаемой при эмболии легочной артерии.
Рефлекс Ларина - это падение артериального давления, вызванное расширением артерий большого круга кровообращения, снижением минутного объема крови в результате возникающей брадикардии и уменьшением ОЦК из-за депонирования крови в печени и селезенке. Кроме того, характерно для рефлекса Ларина появление одышки, связанной с наступающей гипоксией мозга. Полагают, что рефлекс Ларина реализуется за счет усиления тонического влияния п.vagus на сердечно-сосудистую систему при эмболии легочных артерий.
Механизмы компенсации гемодинамических нарушений при хронической сердечной недостаточности
Основным звеном патогенеза ХСН является, как известно, постепенно нарастающее снижение сократительной функции миокарда и падение сердечного выброса. Происходящее при этом уменьшение притока крови к органам и тканям вызывает гипоксию последних, которая первоначально может компенсироваться усиленной тканевой утилизацией кислорода, стимуляцией эритропоэза и т.д. Однако этого оказывается недостаточно для нормального кислородного обеспечения органов и тканей, и нарастающая гипоксия становится пусковым механизмом компенсаторных изменений гемодинамики.
Как и при острой сердечной недостаточности, все эндогенные механизмы компенсации гемодинамических нарушений при ХСН можно подразделить на интракардиальные (механизм Франка - Стерлинга, компенсаторная гиперфункция и гипертрофия миокарда) и экстракарди-альные (разгрузочные рефлексы Бейнбриджа и
Китаева). Такое деление в некоторой степени условно, поскольку реализация как интра-, так и экстракардиальных механизмов находится под контролем нейрогуморальных регуляторных систем.
Экстракардиальные механизмы компенсации функции сердца. В отличие от острой сердечной недостаточности роль рефлекторных механизмов экстренной регуляции насосной функции сердца при ХСН сравнительно невелика, поскольку нарушения гемодинамики развиваются постепенно на протяжении нескольких лет. Более или менее определенно можно говорить о рефлексе Бейнбриджа, который «включается» уже на стадии достаточно выраженной гиперво-лемии.
Особое место среди «разгрузочных» экстракардиальных рефлексов занимает рефлекс Китаева, который «запускается» при митральном стенозе. Дело в том, что в большинстве случаев проявления правожелудочковой недостаточности связаны с застойными явлениями в большом круге кровообращения, а левожелудочковой - в малом. Исключение составляет стеноз митрального клапана, при котором застойные явления в легочных сосудах вызваны не декомпенсацией левого желудочка, а препятствием току крови через левое атриовентрикулярное отверстие - так называемым «первым (анатомическим) барьером». При этом застой крови в легких способствует развитию правожелудочковой недостаточности, в генезе которой рефлекс Китаева играет важную роль.
Рефлекс Китаева - это рефлекторный спазм легочных артериол в ответ на повышение давления в левом предсердии. В результате возникает «второй (функциональный) барьер», который первоначально играет защитную роль, предохраняя легочные капилляры от чрезмерного переполнения кровью. Однако затем этот рефлекс приводит к выраженному повышению давления в легочной артерии - развивается острая легочная гипертензия. Афферентное звено этого рефлекса представлено п.vagus, а эфферентное -симпатическим звеном вегетативной нервной системы. Негативной стороной данной приспособительной реакции является подъем давления в легочной артерии, приводящий к увеличению нагрузки на правое сердце.
Однако ведущую роль в генезе долговременной компенсации и декомпенсации нарушенной
сердечной функции играют не рефлекторные, а вейрогуморальные механизмы, важнейшим из которых является активация симпатоадренало-вой (САС) и ренин-ангиотензин-альдостероновой систем. Говоря об активации САС у пациентов с ХСН, нельзя не указать, что у большинства из них уровень катехоламинов в крови и моче находится в пределах нормы. Этим ХСН отличается от ОСН.
Интракардиальные механизмы компенсации функции сердца. К ним относятся компенсаторная гиперфункция и гипертрофия сердца. Эти механизмы являются неотъемлемыми компонентами большинства приспособительных реакций сердечно-сосудистой системы здорового организма, но в условиях патологии могут превратиться в звено патогенеза ХСН.
Компенсаторная гиперфункция сердца (КГС). КГС выступает как важный фактор компенсации при пороках сердца, артериальной ги-пертензии, анемии, гипертонии малого круга и других заболеваниях. В отличие от физиологической гиперфункции она является длительной и, что существенно, непрерывной. Несмотря на непрерывность, КГС может сохраняться в течение многих лет без явных признаков декомпенсации насосной функции сердца.
Увеличение внешней работы сердца, связанное с подъемом давления в аорте (изометрическая гиперфункция), приводит к более выраженному возрастанию потребности миокарда в кислороде, чем перегрузка миокарда, вызванная повышением объема циркулирующей крови (изотоническая гиперфункция). Иными словами, для осуществления работы в условиях нагрузки давлением мышца сердца использует гораздо больше энергии, чем для выполнения той же работы, связанной с нагрузкой объемом, а следовательно, при стойкой артериальной гипертен-зии гипертрофия сердца развивается быстрее, чем при увеличении ОЦК. Например, при физической работе, высотной гипоксии, всех видах клапанной недостаточности, артерио-венозных фистулах, анемии гиперфункция миокарда обеспечивается за счет увеличения минутного объема сердца. При этом систолическое напряжение миокарда и давление в желудочках возрастают незначительно и гипертрофия развивается медленно. В то же время при гипертонической болезни, гипертензии малого круга, стенозах клапанных отверстий развитие гиперфункции связано с повышением напряжения миокарда при
незначительно измененной амплитуде сокращений. В этом случае гипертрофия прогрессирует достаточно быстро.
Гипертрофия миокарда - это увеличение массы сердца за счет увеличения размеров кардиомиоцитов. Существуют три стадии компенсаторной гипертрофии сердца. Первая, аварийная, стадия характеризуется, прежде всего, увеличением интенсивности функционирования структур миокарда и, по сути, представляет собой компенсаторную гиперфункцию еще не гипертрофированного сердца. Интенсивность функционирования структур (ИФС) - это механическая работа, приходящаяся на единицу массы миокарда. Увеличение ИФС закономерно влечет за собой одновременную активацию энергообразования, синтеза нуклеиновых кислот и белка. Указанная активация синтеза белка происходит таким образом, что вначале увеличивается масса энергообразующих структур (митохондрий), а затем - масса функционирующих структур (мио-фибрилл). В целом увеличение массы миокарда приводит к тому, что ИФС постепенно возвращается к нормальному уровню.
Вторая стадия завершившейся гипертрофии характеризуется нормальной ИФС миокарда и, соответственно, нормальным уровнем энергообразования и синтеза нуклеиновых кислот и белков в ткани сердечной мышцы. При этом потребление кислорода на единицу массы миокарда остается в границах нормы, а потребление кислорода сердечной мышцей в целом увеличено пропорционально возрастанию массы сердца. Увеличение массы миокарда в условиях ХСН происходит за счет активации синтеза нуклеиновых кислот и белков. Пусковой механизм этой активации изучен недостаточно. Считается, что определяющую роль здесь играет усиление трофического влияния симпатоадреналовой системы. Эта стадия процесса совпадает с длительным периодом клинической компенсации. Содержание АТФ и гликогена в кардиомиоцитах также находится при этом в пределах нормы. Подобные обстоятельства придают относительную устойчивость гиперфункции, но вместе с тем не предотвращают исподволь развивающихся в данной стадии нарушений обмена и структуры миокарда. Наиболее ранними признаками таких нарушений являются значительное увеличение концентрации лактата в миокарде, а также умеренно выраженный кардиосклероз.
Третья стадия прогрессирующего кардио-
 |
склероза и декомпенсации характеризуется нарушением синтеза белков и нуклеиновых кислот в миокарде. В результате нарушения синтеза РНК, ДНК и белка в кардиомиоцитах наблюдается относительное уменьшение массы митохондрий, что ведет к торможению синтеза АТФ на единицу массы ткани, снижению насосной функции сердца и прогрессированию ХСН. Ситуация усугубляется развитием дистрофических и склеротических процессов, что способствует появлению признаков декомпенсации и тотальной сердечной недостаточности, завершающейся гибелью пациента. Компенсаторная гиперфункция, гипертрофия и последующая декомпенсация сердца - это звенья единого процесса. Механизм декомпенсации гипертрофированного миокарда включает следующие звенья:
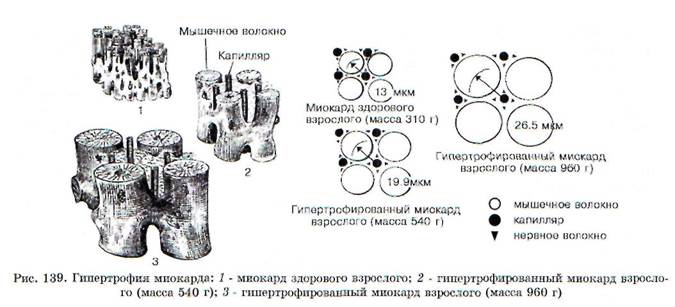
1. Процесс гипертрофии не распространяется на коронарные сосуды, поэтому число капилляров на единицу объема миокарда в гипертрофированном сердце уменьшается (рис. 139). Следовательно, кровоснабжение гипертрофированной сердечной мышцы оказывается недостаточным для выполнения механической работы.
2. Вследствие увеличения объема гипертрофированных мышечных волокон уменьшается удельная поверхность клеток, в связи с этим ухудшаются условия для поступления в клетки питательных веществ и выделения из кардио-миоцитов продуктов метаболизма.
3. В гипертрофированном сердце нарушается соотношение между объемами внутриклеточных
структур. Так, увеличение массы митохондрий и СПР отстает от увеличения размеров миофиб-рилл, что способствует ухудшению энергоснабжения кардиомиоцитов и сопровождается нарушением аккумуляции Ca2t в СПР. Возникает Са2+-перегрузка кардиомиоцитов, что обеспечивает формирование контрактуры сердца и способствует уменьшению ударного объема. Кроме того, Са2*-перегрузка клеток миокарда повышает вероятность возникновения аритмий.
4. Проводящая система сердца и вегетативные нервные волокна, иннервирующие миокард, не подвергаются гипертрофии, что также способствует возникновению дисфункции гипертрофированного сердца.
5. Активируется апоптоз отдельных кардиомиоцитов, что способствует постепенному замещению мышечных волокон соединительной тканью (кардиосклероз).
В конечном итоге гипертрофия утрачивает приспособительное значение и перестает быть полезной для организма. Ослабление сократи-гельной способности гипертрофированного сердца происходит тем скорее, чем сильнее выражены гипертрофия и морфологические изменения в миокарде.
Механизмы декомпенсации сердечной недостаточности
Параллельно с интра- и экстракардиальными компенсаторными изменениями, которые разви-
ваются при сердечной недостаточности, появляются и постепенно прогрессируют повреждения сердечной мышцы, приводящие к снижению ее сократительной способности. На определенной стадии процесса такие явления могут быть обратимыми. При продолжении или усилении действия причинного фактора, вызвавшего сердечную недостаточность, а также при срыве механизмов компенсации развиваются необратимые диффузные изменения миокарда с характерной клинической картиной декомпенсированной сердечной недостаточности.
Патогенез сердечной недостаточности представляется следующим образом. Многочисленный ряд примеров патологии сердечной деятельности (кардиомиопатии, нарушения коронарной перфузии и др.) индуцирует кислородное голодание миокарда. Известно, что в условиях нормального кровоснабжения важным энергетическим субстратом для сердечной мышцы являются свободные жирные кислоты (СЖК), глюкоза и молочная кислота. Гипоксия приводит к нарушению процессов аэробного окисления субстратов в цикле Кребса, к угнетению окисления НАДН в дыхательной цепи митохондрий. Все это способствует накоплению недоокисленных продуктов метаболизма СЖК и глюкозы (ацил-КоА, лактат). Усиленное образование ацил-КоА в кардиомиоцитах негативно сказывается на энергетическом метаболизме клетки. Дело в том, что ацил-КоА является ингибитором аденилат-гранслоказы - фермента, который осуществляет транспорт АТФ из митохондрий в саркоплазму. Аккумуляция ацил-КоА приводит к нарушению этого транспорта, усугубляя энергетический дефицит в клетке.
Единственным источником энергии для кар-диомиоцитов становится анаэробный гликолиз, интенсивность которого в условиях гипоксии резко возрастает. Однако «коэффициент полезного действия» анаэробного гликолиза, по сравнению с эффективностью энергопродукции в цикле Кребса, намного ниже. В силу этого анаэробный гликолиз не в состоянии полностью возместить энергетические потребности клетки. Так, при анаэробном расщеплении одной молекулы глюкозы образуются всего две молекулы АТФ, в то время как при окислении глюкозы до углекислого газа и воды - 32 молекулы АТФ. Нехватка высокоэнергетических фосфатов (АТФ и креатинфосфата) приводит к нарушению энер-
гозависимого процесса удаления ионов кальция из саркоплазмы кардиомиоцитов и возникновению кальциевой перегрузки миокарда.
В норме увеличение [Са2*]1 вызывает образование мостиков между цепочками актина и миозина, что является основой сокращения кардиомиоцитов. Вслед за этим происходит удаление избытка ионов кальция из саркоплазмы и развитие диастолы. Кальциевая перегрузка клеток миокарда при его ишемии ведет к остановке процесса сокращения - расслабления в стадии систолы, формируется контрактура миокарда состояние, при котором кардиомиоциты перестают расслабляться. Возникшая зона асистолии характеризуется повышенным тканевым напряжением, что ведет к сдавлению коронарных сосудов и связанному с этим усугублению дефицита коронарного кровотока.
Ионы Са2+ активируют фосфолипазу А,, которая катализирует расщепление фосфолипидов. В результате этого образуются одна молекула СЖК и одна молекула лизофосфатида. Свободные жирные кислоты обладают детергентоподоб-ным действием и в случае избыточного их накопления в миокарде могут повреждать мембраны кардиомиоцитов. Еще более выраженный кардиотоксический эффект оказывают лизофос-фатиды. Особенно токсичен лизофосфатидилхо-лин, который может провоцировать аритмии. В настоящее время роль СЖК и лизофосфатидов в патогенезе ишемического повреждения сердца никем не оспаривается, однако молекулярная природа необратимого повреждения кардиомиоцитов не сводится только к накоплению этих веществ в клетках сердечной мышцы. Кардио-токсическими свойствами могут обладать и другие продукты метаболизма, например активные формы кислорода.
Активными формами кислорода (АФК) называют супероксидный радикал (02*) и гидро-ксильный радикал НО', которые обладают высокой окислительной активностью. Источником АФК в кардиомиоцитах является дыхательная цепь митохондрий и прежде всего цитохромы, которые в условиях гипоксии переходят в восстановленное состояние и могут быть донорами электронов, «передавая» их молекулам кислорода с образованием не молекулы воды, как это происходит в норме, а супероксидного радикала (02*), Кроме того, образование свободных радикалов катализируется ионами металлов с пере-
менной валентностью (прежде всего, ионами железа), которые всегда присутствуют в клетке. Активные формы кислорода взаимодействуют с молекулами белков и полиненасыщенных жирных кислот, превращая их в свободные радикалы. Вновь образованные радикалы могут, в свою очередь, взаимодействовать с другими молекулами белков и жирных кислот, индуцируя дальнейшее образование свободных радикалов. Таким образом, реакция может принимать цепной и разветвленный характер. Если пероксидации подвергаются белки ионных каналов, то происходит нарушение процессов ионного транспорта. Если гидроперекиси образуются из молекул ферментов, последние теряют свою каталитическую активность.
Образование гидроперекисей полиненасыщенных жирных кислот, входящих в молекулярную структуру мембранных фосфолипидов, способствует изменению биологических свойств мембран. В отличие от жирных кислот гидроперекиси являются водорастворимыми веществами, и появление их в структуре гидрофобного фосфо-липидного матрикса клеточных мембран приводит к формированию пор, пропускающих ионы и молекулы воды. Кроме того, изменяется активность мембраносвязанных ферментов.
Процесс возникновения гидроперекисей жирных кислот является одним из звеньев перекис-ного окисления липидов, которое включает в себя еще свободнорадикальное образование альдегидов и кетонов. Все эти вещества получили название продуктов ПОЛ. Согласно концепции Ф.З. Меерсона, продукты ПОЛ обладают кардиоток-сическими свойствами и накопление их в клетке приводит к повреждению сарколеммы, а также лизосомальных и митохондриальных мембран. На заключительном этапе повреждения, предшествующем гибели клеток, особая роль отводится активации протеолитических ферментов. Обычно эти энзимы находятся в цитоплазме кардиомиоцитов в неактивном состоянии или локализованы внутри лизосом, мембраны которых изолируют их от структурных элементов клетки. В связи с этим в норме протеазы не оказывают цитотоксического действия. В условиях ишемии перегрузка кардиомиоцитов ионами кальция и закисление цитоплазмы за счет накопления лактата приводят к активации внутриклеточных протеаз. Кроме того, повышение проницаемости лизосомальных мембран под дей-
ствием фосфолипаз и продуктов ПОЛ способствует выходу активных протеолитических ферментов в саркоплазму. Конечным звеном этой патогенетической цепочки является некроз кардиомиоцитов в зоне ишемии и их «самопереваривание», которое получило название аутолиза.
Важно отметить, что первыми погибают только кардиомиоциты, отличающиеся высокой интенсивностью энергетического метаболизма и, соответственно, повышенной потребностью в кислороде. В то же время фибробласты и клетки проводящей системы менее зависимы от доставки кислорода и сохраняют свою жизнеспособность. Функциональная активность фиброблас-тов обеспечивает процессы рубцевания.
Клетки проводящей системы, сохраняя жизнеспособность в условиях кислородного голодания, существенно изменяют свои электрофизиологические характеристики, что может способствовать возникновению аритмий. В результате повреждения мембран и снижения образования АТФ изменяется активность К'-, №+-АТФазы, что сопровождается усиленным поступлением натрия в кардиомиоциты и выходом из них калия. Это увеличивает электрическую нестабильность миокарда и способствует развитию аритмий.
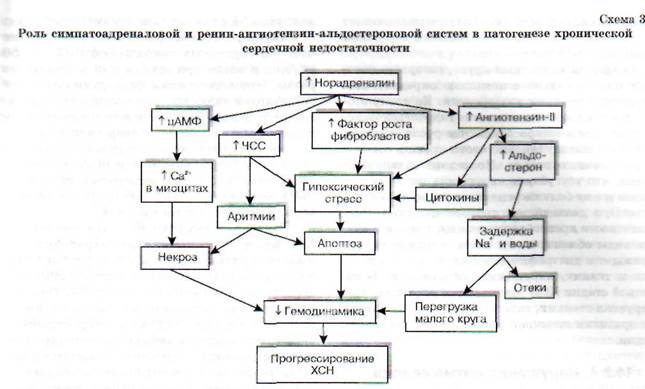
Гипоксическая сократительная дисфункция сердца усугубляется нарушением процессов ней-рогуморальной регуляции функционального состояния миокарда. Сердечные боли, приступы аритмии и другие нарушения являются для организма стрессором, т.е. воздействием чрезмерной силы, на которое организм, как и на любое стрес-сорное воздействие, реагирует активацией сим-патоадреналовой системы. При этом происходит выброс катехоламинов из надпочечников и симпатических нервных терминалей. Однако как и любой другой компенсаторный процесс, активация САС в конце концов приобретает негативную окраску. Наступает период декомпенсации. Схематично последовательность событий представлена на схеме 30.
В настоящее время установлено, что при хронической активации симпатоадреналовой системы происходит постепенная Са2+-перегрузка кардиомиоцитов и их контрактура, нарушается целостность сарколеммы. При гиперактивации ад-ренергической системы формируется электрическая нестабильность миокарда. Последняя способствует возникновению фибрилляции желудоч-
 |
ков сердца, поэтому каждый третий пациент при ХСН погибает внезапно, иногда сердечная смерть наступает на фоне внешнего благополучия и положительной клинической динамики течения ХСН.
Адренергическая тахикардия сопровождается повышением потребности миокарда в кислороде, что наряду с Са2т-перегрузкой еще больше усугубляет энергетический дефицит в клетках миокарда. Включается защитно-приспособительный механизм, получивший название спячки или гибернации кардиомиоцитов. Часть клеток перестает сокращаться и отвечать на внешние стимулы, потребляя при этом минимум энергии и экономя кислород для активно сокращающихся кардиомиоцитов. Таким образом, количество обеспечивающих насосную функцию сердца клеток миокарда может существенно уменьшиться, способствуя усугублению сердечной недостаточности.
Кроме того, гиперактивация симпатоадрена-ловой системы усиливает секрецию ренина почками, выступая в роли стимулятора РААС. Образующийся ангиотензин-И оказывает ряд негативных эффектов на сердечно-сосудистую систему. Он способствует увеличению адренореак-тивности сердца и сосудов, усиливая тем самым
кардиотоксическое действие катехоламинов. Одновременно этот пептид увеличивает периферическое сопротивление кровеносных сосудов, что, безусловно, способствует увеличению постнагрузки на сердце и весьма негативно сказывается на гемодинамике. Кроме того, ангиотен-зин-П может самостоятельно или через активацию образования цитокинов (биологически активные вещества белковой природы, образующиеся в миокарде и других тканях) стимулировать программируемую гибель кардиомиоцитов («апоптоз»).
Наряду с отмеченным повышение уровня ан-гиотензина-П негативно сказывается на состоянии водно-солевого гомеостаза, поскольку этот пептид активирует секрецию альдостерона. В результате в организме задерживается избыточное количество воды и натрия. Задержка натрия повышает осмолярность крови, в ответ на которую происходит активация секреции антидиуретического гормона, что приводит к уменьшению диуреза и еще большей гидратации организма. В итоге повышается ОЦК и увеличивается преднагрузка на сердце. Гиперволемия ведет к раздражению механорецепторов, локализованных в устье полых и легочных вен, «включается» рефлекс Бейнбриджа, возникает рефлектор-
ная тахикардия, что еще больше увеличивает нагрузку на миокард и потребность сердечной мышцы в кислороде.
Создается «порочный круг», разорвать который можно только с помощью определенных фармакологических воздействий. Ко всему этому присоединяется повышение гидростатического давления в микрососудистом русле, что способствует выходу жидкой части крови в ткани и формированию отеков. Последние сдавливают ткани, что усугубляет нарушение микроциркуляции и еще больше усиливает тканевую гипоксию. При дальнейшем прогрессировании недостаточности кровообращения нарушаются и другие виды обмена, в том числе и белковый, что приводит к дистрофическим изменениям в органах и тканях, нарушению их функции. В конечной стадии ХСН развиваются кахексия, маскируемая отеками, гипопротеинемия, появляются признаки почечной и печеночной декомпенсации.
14.3.4. Нарушения ритма сердца
Нарушения сердечного ритма (аритмии) -изменения нормальной частоты, регулярности и источника возбуждения сердца, а также расстройства проведения импульса, нарушения связи и /или последовательности между активацией предсердий и желудочков.
В соответствии с механизмом возникновения аритмий все нарушения сердечного ритма можно условно подразделить на три типа: 1) нарушения автоматизма; 2) нарушения возбудимости; 3) нарушения проводимости. Подобное деление в известном смысле условно потому, что в реальности часто приходится сталкиваться с аритмиями сочетанного характера. Например, при фибрилляции желудочков и предсердий могут иметь место как нарушение возбудимости, так и патология проведения сердечного импульса.
Нарушения сердечного автоматизма
Нарушения сердечного автоматизма - это аритмии, обусловленные нарушением электрофизиологической активности водителей сердечного ритма (синусового и атриовентрикуляр-ного узлов). К этим аритмиям относятся: синусовая брадикардия, синусовая тахикардия, синусовая аритмия, атриовентрикулярная тахикар-
дия, узловой ритм, идиовентрикулярный ритм.
Синусовая брадикардия - это уменьшение частоты сердечных сокращений (ЧСС) до 50 уд./мин и менее при сохранении нормального ритма. Этиологическими факторами синусовой брадикардии являются: повышение тонуса блуждающего нерва, которое может наблюдаться у здоровых людей, чаще - у спортсменов (не требует лечения); первичное поражение синусового узла; повышение внутричерепного давления; гипотиреоз; гипотермия; инфаркт миокарда нижней локализации; передозировка Р-адреноблока-торов или антагонистов кальция.
Синусовая тахикардия - это повышение ЧСС более 100 уд./мин при сохранении нормального ритма. Этиологические факторы: нормальная реакция на различные стрессорные факторы (волнение, беспокойство, страх, физическая нагрузка); патологические состояния, в частности - лихорадка, гипотония, тиреотоксикоз, анемия, гиповолемия, эмболия легочной артерии, ишемия миокарда, сердечная недостаточность, шок, митральный стеноз; прием некоторых лекарств (атропин, катехоламины, тиреоидные препараты) или некоторых биологически активных веществ (алкоголь, никотин, кофеин).
Синусовая аритмия - это периодически сменяющие друг друга эпизоды синусовой тахикардии или брадикардии при сохранении синусовой импульсации. По данным ЭКГ, комплекс QRS обычно не деформирован, интервалы R-R укорочены или удлинены, но равны. В норме она может быть следствием периодического изменения тонуса блуждающего нерва, так называемой дыхательной аритмией (повышение ЧСС при вдохе и снижение на выдохе). Этиологические факторы: эмоциональный стресс, климакс, тиреотоксикоз, миокардит.
Узловой ритм - это нарушение, при котором роль водителя ритма берет на себя атрио-вентрикулярный узел. При этой патологии ЧСС снижается до 40-60 уд./мин. Причинами подобного нарушения автоматизма наиболее часто являются интоксикация, которая приводит к слабости синусового узла, или блокада внутри-предсердного проведения импульса.
Атриовентрикулярные - реципрокные па-роксизмальные тахикардии - нарушения ритма, связанные с повышенной возбудимостью атриовентрикулярного узла (АВ-узла). Эта группа нарушений ритма составляет 85% всех
Часть III. ПАТОФИЗИОЛОГИЯ ОРГАНОВ И СИСТЕМ
наджелудочковых аритмий. Электрофизиологический механизм данных аритмий представляет собой сочетание нарушения автоматизма и патологии проведения импульса (re-entry). Этиология АВ-реципрокных пароксизмальных тахикардии остается до сих пор неизвестной, но почти у 1/3 всех пациентов, страдающих этим типом нарушений ритма, приступы сердцебиения связаны с психоэмоциональной нагрузкой.
Идиовентрикулярный ритм - это нарушение, при котором роль водителя ритма берут на себя ножки пучка Гиса или волокна Пур-кинье. Ритм при этом урежается до 10-30 уд./ мин. Такое нарушение автоматизма развивается при повреждении синусового и атриовентрику-лярного узлов и ведет к нарушению центральной гемодинамики, что может закончиться гибелью пациента.
Нарушения возбудимости сердца
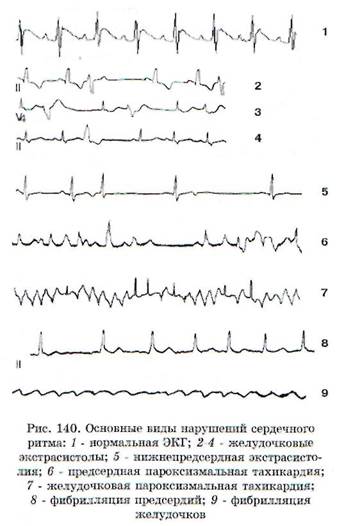
Нарушения возбудимости сердца лежат в основе таких видов аритмий, как экстрасистолии, желудочковые тахикардии, полиморфная желудочковая тахикардия, трепетание желудочков и предсердий, фибрилляция желудочков и предсердий, внезапная остановка сердца. Основные из них представлены на рис. 140.
Экстрасистолия - внеочередное сокращение сердца. Экстрасистолы, исходящие из синусового узла, называются номотопными. Однако и в этом случае источником экстрасистолы являются не клетки водителя ритма (пейсмекера), а расположенные в его окружении клетки, обладающие латентным автоматизмом, но не проявляющие пейсмекерной активности в нормальных условиях. Пейсмекерной активностью называют способность клеток к спонтанной деполяризации. Обычно пейсмекерная активность АВ-узла, ножек пучка Гиса и волокон Пуркинье подавляется импульсами, поступающими из синусового узла, но в условиях блокады проведения импульсов от предсердий к желудочкам сердца эти латентные пейсмекеры могут активироваться и вызывать появление экстрасистол. Ге-теротопные экстрасистолы исходят из любого участка проводящей системы, исключая синусовый узел. Эктопические экстрасистолы имеют источник внеочередного возбуждения, локализующийся в миокарде за пределами проводящей системы сердца. Аналогичная ситуация ча-
сто складывается в очаге ишемии при инфаркте миокарда. В зависимости от локализации эктопического очага различают предсердные, атрио-вентрикулярные, левожелудочковые, правоже-лудочковые и перегородковые экстрасистолы.
Одиночные экстрасистолы не вызывают серьезных расстройств гемодинамики и клинически проявляются ощущением «перебоев» в работе сердца. Однако множественные и особенно по-литопные экстрасистолы, т.е. исходящие из нескольких эктопических центров, могут вызвать серьезные нарушения гемодинамики по двум причинам. Во-первых, многие экстрасистолы гемодинамически малоэффективны, поскольку процесс внеочередного сокращения может возникнуть в период, когда сердце еще не успело полностью расслабиться и, следовательно, конечный диастолический объем желудочков в этот момент остается сниженным, так же как и ударный объем. Во-вторых, после экстрасистолы следует компенсаторная пауза, т.е. удлиненная диастола, в период которой миокард находится в состоянии рефрактерности и не чувствителен к импульсу, поступающему из синусового узла. Наиболее выраженные нарушения гемодинамики наблюдаются при желудочковых экстрасистолах.
Желудочковые экстрасистолы (ЖЭ) - преждевременные желудочковые сокращения, обусловленные наличием очага автоматизма в желудочках. Этиологические факторы ЖЭ: ИБС и ее осложнения (в частности, острый инфаркт миокарда), кардиомиопатии, нарушения электролитного и кислотно-щелочного баланса, гипоксия, эндокринные заболевания (тиреотоксикоз), инфекции, прием некоторых лекарств (сердечные гликозиды и антиаритмические средства). ЖЭ могут регистрироваться и у практически здоровых людей. Так, например, по данным суточного мониторирования ЭКГ, ЖЭ отмечаются в 70-80% случаев у лиц в возрасте старше 60 лет, причем наиболее часто обнаруживаются бессимптомные ЖЭ.
Электрокардиографически ЖЭ характеризуются (рис. 140). появлением преждевременных комплексов QRS, отличающихся от нормальных комплексов шириной более 0,12 с, деформацией, наличием предшествующего укороченного интервала R-R (рис. 141). Зубец Т, как правило, увеличен и так же, как сегмент S-T, расположен дискордантно, т.е. направлен в другую сторону
 |
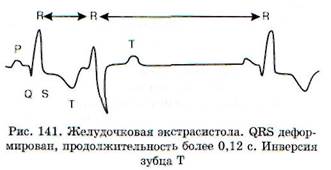 |
по отношению к самому высокоамплитудному зубцу комплекса QRS. Экстрасистолическому комплексу QRS не предшествует зубец Р. Клинически ЖЭ проявляются как ощущение сердцебиения или дискомфорт в груди, ощущение перебоев в работе сердца (в связи с наличием компенсаторных пауз после ЖЭ).
Среди желудочковых экстрасистол наиболее часто встречаются единичные ЖЭ, реже множественные ЖЭ, парные ЖЭ, бигеминия (состояние, когда каждой нормальной систоле сопутствует ЖЭ), политопные ЖЭ (возникающие из разных отделов миокарда). Появление парных ЖЭ увеличивает риск смерти. Особую опасность представляют ранние желудочковые экстрасистолы, при которых эктопический импульс приходится на так называемую «ранимую фазу сер-
дечного цикла». Ранимая фаза сердечного цикла - это интервал времени, когда процесс репо-ляризации еще полностью не завершился, сердце находится в состоянии относительной рефрак-терности и любой экстрастимул, в том числе эктопический импульс, имеющий желудочковую локализацию, может вызвать появление не только желудочковой экстрасистолы, но и желудочковой фибрилляции, которая может закончиться гибелью пациента. Электрокардиографически ранимая фаза почти полностью соответствует зубцу Т, поэтому подобные экстрасистолы называют «экстрасистола R на Т» (R на Т). Появление ранних ЖЭ является негативным прогностическим признаком, поскольку часто предшествует внезапной сердечной смерти.
Мерцательная аритмия (фибрилляция предсердий) - это отсутствие скоординированных сокращений предсердий, которое электрокардиографически характеризуется исчезновением зубца Р. Фибрилляция предсердий приводит к прекращению гемодинамически эффективных сокращений предсердий. Она проявляется нерегулярными мелкими колебаниями предсердий различной амплитуды и формы с частотой 350-600 в минуту, которые не удается зарегистрировать на обычном электрокардиографе. Желудочковые сокращения также нерегулярны. Различают тахисистолическую (ЧСС более 100 уд./ мин), нормосистолическую (ЧСС = 60-90 уд./мин) и брадисистолическую (ЧСС ниже 60 уд./мин) формы фибрилляции предсердий. Этиологические факторы: атеросклероз, гипотония, кардио-миопатия и ревматические заболевания сердца, тиреотоксикоз, иногда видимая причина отсутствует. При тахиаритмической форме мерцательной аритмии частота желудочковых сокращений может достигать 150-240 в минуту, причем во время пароксизма может развиться резкая ги-
 492
492
потония или отек легких вследствие перегрузки сердца и острой левожелудочковой недостаточности.
Трепетание предсердий - нарушение процессов возбуждения и проведения в предсердиях, которое электрокардиографически характеризуется исчезновением зубца Р и появлением вместо него частых низкоамплитудных колебаний, так называемых зубцов F, которые получили свое название от английского слова flatter - колебание. При этой патологии частота сокращений предсердий составляет более 220 в минуту, а желудочков - 120-180 в минуту. Одновременно возникают блокады АВ-проведения 1:1, 2:1, 3:1, 4:1 и даже 5:1, причем возможны постепенные или быстрые переходы. Комплексы QRS нормальные, реже напоминают желудочковые экстрасистолы. Различают тахи-, нормо- и брадисистолическую формы трепетания предсердий. Этиологические факторы те же, что и при мерцательной аритмии.
Желудочковая тахикардия (ЖТ) - частый и в основном регулярный ритм, берущий свое начало: а) в сократительном миокарде желудочков; б) в сети Пуркинье; в) в ножках пучка Гиса. Электрокардиографически ЖТ характеризуется появлением серии из трех и более ЖЭ с дискордантно расположенным сегментом S-T по отношению к основному отклонению комплекса QRS. Интервалы R-R могут быть регулярными или различаться по продолжительности. Часто ЖТ представлена комплексом полиморфных ЖЭ. Большинство ЖТ в своей основе обусловлено механизмом re-entry с локализацией критического участка циркуляции электрического возбуждения в субэндокардиальной области. В более редких случаях ЖТ возникает вследствие нарушения автоматизма. Чаще всего ЖТ носит пароксизмальный характер, но иногда имеет место стабильная многочасовая ЖТ.
Причинами ЖТ, как правило, являются тяжелые заболевания миокарда: хроническая ишемия и острый инфаркт миокарда, постинфарктный кардиосклероз, миокардиты, миокардиопа-тии, ревматические клапанные пороки сердца, синдром WPW, тяжелая сердечная недостаточность различной этиологии, синдром удлиненного интервала Q-T. Реже причинами ЖТ могут быть: тиреотоксикоз, гипоксемия, нарушение кислотно-щелочного баланса, гипокалиемия, интоксикация препаратами наперстянки, хини-
дином, новокаинамидом, катехоламинами, циклопропаном.
Среди различных нарушений сердечного ритма желудочковая тахикардия занимает особое место, поскольку может привести к перегрузке сердца или перейти в фибрилляцию желудочков (ФЖ). Первое из этих осложнений чревато развитием острой левожелудочковой недостаточности, а второе - прекращением кровоснабжения жизненно важных органов и гибелью пациента. Именно поэтому появление стойкой ЖТ повышает риск внезапной сердечной смерти в 5-6 раз по сравнению с пациентами, не имеющими желудочковых аритмий.
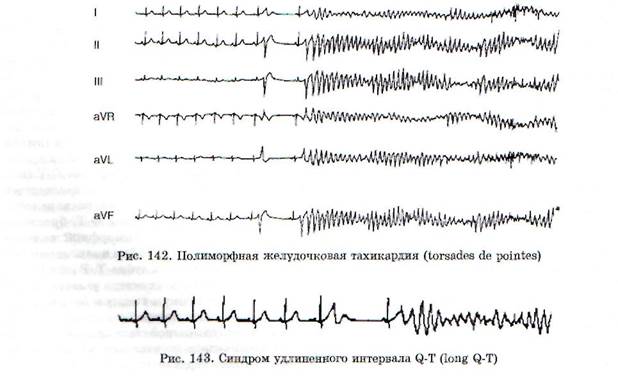
Полиморфная (пароксизмальная) желудочковая тахикардия в большинстве случаев возникает в виде пароксизмов с частотой более 200 уд./мин. Обычно развивается вследствие неконтролируемой терапии антиаритмическими средствами, а также как проявление врожденного синдрома удлиненного интервала Q-T. Электрокардиографическая картина полиморфной ЖТ представлена рис. 142, на котором видно, что желудочковые комплексы как бы «вьются» вокруг изоэлектрической оси. Появлению этой аритмии предшествуют брадикардия и удлинение интервала Q-T. Полиморфная ЖТ развивается по механизму триггерного автоматизма (см. ниже) и обычно носит обратимый характер. Однако по сравнению с мономорфной ЖТ эта аритмия значительно чаще трансформируется в ФЖ.
Причинами развития этой опасной для жизни аритмии могут быть: гипокалиемия, интоксикации, миокардит, ишемия, некоторые лекарственные средства и комбинация факторов. В частности, она может развиться даже при приеме антиаритмических препаратов (хинидин, новокаинамид, амиодарон, соталол и др.).
Синдром удлиненного интервала Q-T (long Q-T) может быть приобретенным и наследственным. Электрокардиографически он характеризуется удлинением интервала Q-T, брадикар-дией, возникновением полиморфной желудочковой тахикардии (рис. 143) и появлением волны U, следующей после зубца Т. Волну U из-за маленькой амплитуды не всегда удается зарегистрировать. Клинически синдром long Q-T проявляется внезапной потерей сознания и возникновением желудочковой тахикардии, которая может закончиться спонтанным восстановлением нормального сердечного ритма или наоборот,
перейти в желудочковую фибрилляцию с нарушением центральной гемодинамики и гибелью пациента.
Приобретенный синдром связан с употреблением некоторых лекарственных препаратов, врожденный - с мутациями генов, кодирующих структуру полипептидной цепочки быстрого Ыа+-канала или двух типов К+- каналов. Известно, что деполяризация кардиомиоцитов начинается с быстрой активации Ма+-каналов, которая сменяется такой же быстрой инактивацией этих каналов. Весь цикл занимает несколько миллисекунд. Мутация гена, кодирующего белок Ма*-канала, приводит к замедлению процесса инактивации этого канала. В результате возникает перегрузка кардиомиоцитов ионами Na+, тормозится процесс восстановления нормального градиента ионов и замедляется реполяриза-ция кардиомиоцитов. Эти события могут индуцировать появление желудочковых аритмий по механизму ранней постдеполяризации и проявляются на ЭКГ удлинением интервала Q-T.
Как известно, процесс реполяризации обеспечивается К*-каналами, которые при этом открываются. В настоящее время идентифицированы два гена, мутация которых приводит к инактивации этих каналов, что ведет к замедле-
нию реполяризации. Наследственная форма синдрома long Q-T встречается достаточно редко.
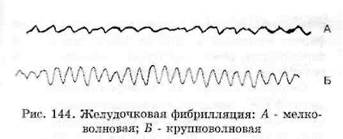
Фибрилляция (и трепетание) желудочков -это хаотическое асинхронное возбуждение отдельных мышечных волокон или небольших групп волокон с остановкой сердца и прекращением кровообращения. Эти аритмии представляют наибольшую опасность, так как они в отсутствие экстренных мероприятий в течение 3-5 мин могут привести к летальному исходу. Электрокардиологически фибрилляция желудочков характеризуется появлением волн низкой амплитуды (менее 0,2 мВ) и различной формы с частотой от 300 до 600 в минуту. Такую ФЖ называют мелковолновой ФЖ (рис. 144). Трепетание желудочков характеризуется на ЭКГ появлением волн с нерегулярными большими ос-цилляциями при частоте 150-300 в минуту. Трепетание желудочков нередко называют крупноволновой ФЖ. И при той, и при другой форме ФЖ невозможно выделить комплекс QRS, сегмент S-T и зубец Т. Возникает фибрилляция желудочков при различных сердечно-сосудистых заболеваниях, особенно часто при острой коронарной недостаточности, ишемии миокарда, а также при тяжелом течении кардиомиопатии.
Следует особо отметить, что желудочковые


 |
аритмии имеют тенденцию трансформироваться в более тяжелые формы, например множественные желудочковые экстрасистолы в пароксиз-мальную тахикардию, а последняя - в фибрилляцию сердца, которая может закончиться асистолией и внезапной сердечной смертью.
Внезапная остановка сердца может быть двух типов: а) асистолия желудочков, когда отсутствуют и сокращения желудочков, и их биоэлектрическая активность; б) электромеханическая диссоциация (ЭМД) - крайне опасное состояние сердца, когда на ЭКГ регистрируется электрическая активность при отсутствии эффективного сокращения миокарда.
Причиной внезапной остановки сердца могут быть ИБС, тромбоэмболия легочных артерий, гипертрофия миокарда и кардиомиопатия, первичная или вторичная легочная гипертония, сердечная недостаточность, миокардиты, пороки сердца, синдром удлиненного интервала Q-T и ряд других заболеваний. Феномен электромеханической диссоциации развивается при ишемии миокарда, если она сопровождается выраженным нарушением внутриклеточного транспорта Са2* на уровне СПР при сохраненной активности Na+-, К*-АТФазы сарколеммы. В результате возникающий потенциал действия не влечет за собой сокращения миокарда, что обычно заканчивается гибелью пациента.
Внезапная коронарная смерть может наступить в любом возрасте, в том числе в молодом и даже в детском. По данным ВОЗ, частота ВСС составляет 30 случаев в неделю на 1 млн населения, или около 12% всех случаев естественной смерти. В старших возрастных группах внезапная коронарная смерть наступает на фоне выраженных атеросклеротических изменений коронарных артерий, часто до этого клинически не проявлявшихся, а также на фоне бессимптомной ИБС. Непосредственными причинами внезапной сердечной смерти являются в основном фибрилляция желудочков и желудочковая та-
хикардия, а также асистолия или резкая бради-кардия (на их долю приходится около 20% случаев).
Таким образом, внезапная остановка сердца, о которой мы говорили выше, является только одной из причин ВСС. Последняя наступает или мгновенно, или в течение 2 ч после появления первых симптомов коронарной катастрофы у негоспитализированных больных, имевших до этого заболевания сердца, но находившихся, с точки зрения врача, в относительно стабильном, неопасном для жизни состоянии. На вскрытии у таких пациентов не удается выявить признаки острого инфаркта миокарда. Фатальные нарушения ритма чаще развиваются на фоне электрической нестабильности миокарда, возникающей у больных с морфологическими изменениями в сердце. Однако ВСС возможна и при отсутствии изменений структуры сердца. Причиной ВСС в этом случае являются так называемые идиопатические аритмии, т.е. нарушения ритма неясной этиологии. Например, идиопатические ЖФ составляют приблизительно 1% всех случаев остановки сердца во внегоспитальных условиях. Причиной таких аритмий может явиться стресс-индуцированная электрическая нестабильность сердца (Б. Лаун).
Нарушения проводимости включают в себя поперечную блокаду сердца, блокаду правой и/ или левой ножек пучка Гиса, синдром Вольфа-Паркинсона-Уайта.
Поперечная блокада - это нарушение проведения возбуждения в области атриовентри-кулярного узла. Поперечная блокада сердца, в свою очередь, подразделяется на блокаду I, II, III и IV степени. Первые три степени называют еще неполной, а последнюю - полной поперечной блокадой сердца.
Поперечная блокада I степени проявляется задержкой проведения импульса в АВ-узле. Электрокардиографически она характеризуется удлинением интервала P-Q. Это расстройство сердечного ритма не сказывается на гемодинамике и чаще всего является следствием усиления вагусных влияний на миокард или результатом интоксикации сердечными гликозидами.
Поперечная блокада II степени характеризуется тем, что в структуре каждого последую-
 щего ЭКГ-цикла интервал P-Q удлиняется все больше и больше до тех пор, пока не происходит выпадения одного желудочкового комплекса (период Самойлова-Венкенбаха), после чего продолжительность интервала P-Q возвращается к норме, но тут же вновь начинает удлиняться. Таким образом, процесс носит циклический характер. Возникновение периодов Самойлова-Венкенбаха связано с формированием сначала относительной, а затем абсолютной рефрактер-ности АВ-узла. В последнем случае АВ-узел оказывается неспособным к проведению возбуждения от предсердий к желудочкам. Очередное сокращение желудочков выпадает. В течение этой паузы возбудимость АВ-узла восстанавливается до нормы, и весь цикл повторяется вновь. Клинически этот вид блокады проявляется ощущением «перебоев в работе сердца». Это расстройство проводимости не влияет на гемодинамику и также является следствием усиления тонической активности п.vagus или результатом интоксикации сердечными гликозидами.
щего ЭКГ-цикла интервал P-Q удлиняется все больше и больше до тех пор, пока не происходит выпадения одного желудочкового комплекса (период Самойлова-Венкенбаха), после чего продолжительность интервала P-Q возвращается к норме, но тут же вновь начинает удлиняться. Таким образом, процесс носит циклический характер. Возникновение периодов Самойлова-Венкенбаха связано с формированием сначала относительной, а затем абсолютной рефрактер-ности АВ-узла. В последнем случае АВ-узел оказывается неспособным к проведению возбуждения от предсердий к желудочкам. Очередное сокращение желудочков выпадает. В течение этой паузы возбудимость АВ-узла восстанавливается до нормы, и весь цикл повторяется вновь. Клинически этот вид блокады проявляется ощущением «перебоев в работе сердца». Это расстройство проводимости не влияет на гемодинамику и также является следствием усиления тонической активности п.vagus или результатом интоксикации сердечными гликозидами.
Поперечная блокада III степени выражается в том, что через АВ-узел проходит от предсердий к желудочкам только каждый второй или третий импульс. Частота сердечных сокращений значительно урежается, поэтому могут возникать серьезные нарушения гемодинамики.
Полная поперечная блокада - это состояние проводимости, при котором ни один импульс не проходит от предсердий к желудочкам. Предсердия при этом сокращаются в синусовом ритме, а желудочки - в идиовентрикулярном. Возникает выраженная брадикардия, которая вызывает тяжелые нарушения центральной гемодинамики, сопровождающиеся нарушением кровоснабжения головного мозга и эпизодами потери сознания продолжительностью от нескольких секунд до нескольких минут (синдром Морга-ньи - Эдемса - Стокса). Этот синдром опасен тем, что может закончиться гибелью пациента в результате асистолии. Единственным эффективным способом лечения этой патологии является имплантация искусственного водителя ритма.
Блокада правой и/или левой ножки пучка Гиса - опасное нарушение проведения импульсов по одной из ножек пучка Гиса. Опасность заключается в том, что при этой блокаде происходит асинхронное сокращение желудочков, что ведет к уменьшению ударного объема и развитию сердечной недостаточности. Это расстрой-
ство наиболее часто является результатом инфаркта миокарда в области межжелудочковой перегородки, реже - следствием ревматической гранулемы и других заболеваний сердца.
Синдром Вольфа - Паркинсона - Уайта (синдром WPW, синдром преждевременного возбуждения). Отличительной чертой этого синдрома является то, что возбуждение к желудочкам приходит двумя путями: а) через АВ-узел и б) по так называемому пучку Кента (аномальный дополнительный путь проведения импульса между предсердиями и желудочками). При этом происходит взаимное наложение проводимых импульсов и в 50% случаев возникает желудочковая тахиаритмия. Как известно, в норме волна возбуждения из синусного узла распространяется по предсердиям и достигает ат-ривентрикулярного узла, где происходит задержка проведения импульса (атриовентрикуляр-ная задержка), поэтому желудочки сокращаются после предсердий с небольшим опозданием. Однако у пациентов с синдромом WPW между предсердиями и желудочками имеется дополнительный путь проведения - пучок Кента, по которому импульс проходит без всякой задержки. По этой причине желудочки и предсердия могут сокращаться одновременно, что ведет к нарушению внутрисердечной гемодинамики и снижает эффективность насосной функции сердца.
Кроме того, опасность представляет и столкновение импульса из атриовентрикулярного узла с волной возбуждения, поступившей в желудочек по пучку Кента. Это может вызвать появление желудочковой экстрасистолы (внеочередного сокращения желудочка сердца). Бели импульс поступит из АВ-узла в тот момент, когда желудочки находятся в фазе относительной рефрак-терности, т.е. тогда, когда процесс реполяриза-ции еще полностью не завершен, то желудочковая экстрасистола может индуцировать появление желудочковой тахикардии или даже фибрилляции. В силу этого период относительной рефрактерности получил название ранимой фазы сердечного цикла. На ЭКГ этот период соответствует зубцу Т.
Выделяют три основных электрокардиографических признака синдрома WPW: а) укороченный интервал P-R на фоне синусового ритма; б) «растянутый» сверх нормы комплекс QRS с пологой начальной частью; в) вторичные изменения сегмента S-T, при которых зубец Т на-


 правлен дискордантно (в обратном направлении) по отношению к комплексу QRS.
правлен дискордантно (в обратном направлении) по отношению к комплексу QRS.
Факторы, приводящие к нарушениям сердечного ритма
Все причины многочисленных тахи- и бради-аритмий можно условно подразделить на четыре группы: 1) нарушения нейрогенной и эндокринной (гуморальной) регуляции электрофизиологических процессов в специализированных или сократительных клетках сердца; 2) органические поражения миокарда, его аномалии, врожденные или наследственные дефекты с повреждением электрогенных мембран и клеточных структур; 3) сочетание нарушений нейрогумо-ральной регуляции ритма и органических заболеваний сердца; 4) аритмии, вызванные лекарственными препаратами. Таким образом, практически любое заболевание кровеносной системы может осложниться нарушениями сердечного ритма. Однако в данном разделе рассматриваются только аритмии, связанные с нарушениями нейрогуморальной регуляции сердечного ритма или с употреблением некоторых лекарственных препаратов.
Нарушения нейрогенной и эндокринной регуляции электрофизиологических процессов в кардиомиоцитах и клетках проводящей системы сердца. Одной из основных причин нарушений сердечного ритма и проводимости является изменение физиологического соотношения между тонической активностью симпатических и парасимпатических элементов, иннервирующих сердце. Важно отметить, что повышение тонической активности симпатического звена вегетативной нервной системы способствует возникновению аритмий, в то время как стимуляция п.vagus, как правило, повышает электрическую стабильность сердца.
Описаны расстройства сердечного ритма, связанные с заболеваниями головного мозга, особенно часто с нарушениями мозгового кровообращения. Большой интерес вызывают спонтанные, психогенные по своей природе аритмии у больных неврозами, психопатиями, вегетативной дистонией. Число аритмий психосоматического генеза в наше время увеличивается.
В эксперименте на животных практически любую из известных форм аритмий - от простой синусовой тахикардии до фибрилляции желудочков - можно вызвать, воздействуя на некоторые
отделы головного мозга: кору, лимбические структуры и в особенности гипоталамо-гипофи-зарную систему, с которой тесно связаны находящиеся в ретикулярной формации продолговатого мозга центры симпатической и парасимпатической регуляции сердечной деятельности. Одним из наиболее ярких примеров нарушений ритма, обусловленных дисбалансом симпатического и парасимпатического звеньев вегетативной нервной системы, является снижение электрической стабильности сердца при психоэмоциональном стрессе. По данным P.Reich et al. (1981), психологический стресс в 20-30% случаев предшествует появлению угрожающих жизни сердечных аритмий. Патогенез стресс-индуцированных аритмий весьма сложен и до конца неясен. Вполне возможно, что он связан с прямым воздействием катехоламинов на миокард. Вместе с тем известно, что высокие концентрации адреналина в крови, активируя р-адренорецепторы почечных канальцев, способствуют усилению экскреции К1 и развитию гипокалиемии. Последняя вызывает нарушения процессов реполяризации, создавая условия для развития самих опасных желудочковых тахиаритмий, в том числе желудочковой фибрилляции и внезапной сердечной смерти. Фармакологическая или хирургическая симпатэктомия устраняет влияние различных типов стресса на ритм сердца и повышает электрическую стабильность миокарда. Такой же эффект оказывает и стимуляция блуждающего нерва, которая способствует угнетению высвобождения норадреналииа из окончаний симпатических нервов и ослаблению адренореактив-ности сердца.
Говоря о роли эндокринных нарушений в патогенезе аритмий, следует указать, что избыточная продукция тиреоидных гормонов способствует увеличению количества адренорецепторов в миокарде и повышению их чувствительности к эндогенным катехоламинам. По этой причине у больных тиреотоксикозом, как правило, наблюдаются тахикардия и нарушения сердечного ритма, обусловленные повышением адренореак-тивности сердца. Одной из частых «эндокринных» причин нарушений электрической стабильности сердца является избыточное образование минералокортикоидов в коре надпочечников (первичный и вторичный альдостероиизм). Реже аритмии возникают при гиперсекреции глюко-кортикоидных гормонов (болезнь и синдром
Иценко - Кушинга) или длительном приеме их фармакологических аналогов.
Механизм аритмогенного эффекта минерало-кортикоидов и, прежде всего, наиболее активного из них - альдостерона - связан с дисбалансом Na"/K+B организме. Альдостерон, действуя на почечные канальцы, вызывает задержку в организме Na* и усиление экскреции К', в результате чего возникает гипокалиемия, которая способствует нарушению процессов реполяриза-ции и возникновению аритмий по триггерному механизму (см. ниже). Умеренное аритмогенное влияние глюкокортикоидов обусловлено тем, что природные (гидрокортизол, кортизол, кортико-стерон) и синтетические (преднизолон, дексаме-тазон) гормоны этой группы не являются «чистыми» глюкокортикоидами, они обладают слабым сродством к рецепторам альдостерона в почечных канальцах. Именно этим свойством объясняется способность данных биологически активных веществ провоцировать аритмии у пациентов, получающих их длительное время.
Аритмии, вызванные лекарственными препаратами. Часто причиной аритмий являются лекарственные препараты, обладающие собственной аритмогенной активностью. В первую очередь это относится к сердечным гликозидам и диуретикам. Мочегонные препараты, усиливая экскрецию калия, способствуют возникновению гипокалиемии. Сердечные гликозиды (дигиталис и др.) имеют свойство накапливаться в организме, ингибируя при этом NaH'-, К+-АТФазу, локализованную на сарколемме кардиомиоцитов. Снижение активности этого фермента сопровождается снижением уровня Кт и увеличением концентрации NaT в саркоплазме. Накопление натрия в цитоплазме кардиомиоцитов приводит к усилению Ыа+/Са2+-обмена, что сопровождается активным поступлением Са2' в клетки миокарда и способствует усилению насосной функции сердца. Однако при этом формируется Са2+-пере-грузка кардиомиоцитов. Кроме того, снижение внутриклеточной концентрации К* вызывает замедление процессов реполяризации и тем самым способствует возникновению ранних деполяризаций и аритмий по механизму триггерно-го автоматизма.
Лекарственные аритмии могут быть вызваны и антиаритмическими препаратами. У больных ХСН, длительное время получавших блокаторы Ыа+-каналов (флекаинид, этацизин и др.) или
блокатор К+-каналов D-соталол, повышается частота случаев внезапной сердечной смерти и сокращается общая продолжительность жизни. Было установлено, что D-соталол ингибирует К'-каналы, что ведет к замедлению процесса реполяризации, возникновению ранних реполяризации и опасных желудочковых аритмий по механизму триггерного автоматизма. Механизм аритмогенного действия блокаторов Na'-каналов у пациентов с ХСН неизвестен.
Патогенез нарушений сердечного ритма
Следует выделить два основных механизма нарушений ритма сердечных сокращений: 1) патологию образования импульса (нарушения автоматизма и повышение возбудимости) и 2) дефекты проведения импульс