2018-01-21
2018-01-21 1517
1517
9.1 Ацетил-коа, його характеристика й біогенез
Багато мікробних метаболітів, незважаючи на гадану складність їхніх структур, побудовані з декількох структурних одиниць. До їхнього числа відносяться так названі ацетатні одиниці. Вони є метаболічними попередниками кислот циклу Кребса, беруть участь у біосинтезі ліпідів, а також численних продуктів, які прийнято відносити до вторинних метаболітів. Закономірності включення ацетату в ті або інші синтетичні реакції визначаються генетичними особливостями культури, наявністю відповідних ферментних систем, специфікою в прояві їхньої активності, фізіологічним станом клітини. У реакціях синтезу оцтова кислота виступає звичайно у вигляді своєї КоА-похідної – ацетил-КоА. Метаболічним попередником ацетил-КоА є піровиноградна кислота. Рідше, як вторинний процес, вона утворюється з жирних кислот у результаті β-окислення.
Ацетил-КоА входить до метаболічного фонду клітин. Пул ацил-КоА в 20-30 разів вище у високопродуктивних штамів, чим у низькопродуктивних, якщо в синтезі структур цільових продуктів беруть участь активовані ацилпохідні. Специфічна активність ацил-КоА синтетаз у високо- і низькопродуктивних штамів, як правило, не відрізняється. Існує більше підстав вважати, що пул ацил-КоА похідних проявляє регуляторну функцію відносно тіоестераз, тобто ферментів, які руйнують тіоефірний зв'язок між коензимом А та ацильним угрупуванням.
Про потенційну можливість синтезу через ацил-КоА можна стверджувати по вмісту в середовищі піровиноградної та молочної кислот. Їх наявність може свідчити про те, що окислювальне декарбоксилування піровиноградної кислоти обмежене.
Відомо декілька шляхів утворення ацетил-КоА в організмах. Одним з джерел може служити піровиноградна кислота. Утворення ацетил-КоА з піровиноградної кислоти проходить ряд стадій. Відомо, що в декарбоксилуванні піровиноградної кислоти приймає участь тіамінпірофосфат. В цій системі кетокислота в незворотній реакції приєднується до вуглецевого атому тіазольного кільця тіамінпірофосфату з утворенням похідної α-оксикислоти. Остання сполука далі декарбоксилюється з утворенням альдегіду і тіамінпірофосфату. У специфічному випадку спочатку з пірувату утворюється α –лактил-2´-тіамінпірофосфат («активований піруват»), а потім α-оксиетил-2´-тіамінпірофосфат («активований ацетальдегід»), який потім переноситься на ліпоєву кислоту.
Утворення ацетилліпоєвої кислоти представляє собою окислювально-відновну реакцію, в якій ліпоєва кислота відновлюється, а ацетальдегід окислюється. Потім у результаті переетерифікації ацетильна група переноситьсяз ацетилліпоєвої кислоти на коензим А. Відновлена ліпоєва кислота знову окислюється в дисульфідну форму при участі дегідрогенази. Усі ферменти, що беруть участь в цьому ланцюгу реакцій утворюють мультиферментний комплекс. Ліпоєва кислота приєднується до ферментного білку ациламідним зв’язком.
У реакціях синтезу ацетил-КоА може безпосередньо приймати участь оцтова кислота. В його утворенні беруть участь ацетат, АТФ і КоА. У більшості бактерій активація оцтової кислоти здійснюється через ацетилфосфат, а майже у всіх інших організмів через ацетиладенілат. В обох випадках синтез відбувається в дві стадії, проміжними продуктами відповідно виступають ацетилфосфат й ацетиладенілат.
Суттєвою особливістю всіх перерахованих реакцій є утворення тіоефіру. Відомо, що тіоефіри більш активні, чим їхні кисневі аналоги, тому що на відміну від тіоефірів вони добре стабілізовані резонансом. Угруповання С-О здобуває карбонільний характер, внаслідок чого атом кисню має негативний заряд, атом вуглецю – позитивний.
9.2 Малоніл-КоА
В якості нуклеофільної сполуки ацетил-КоА проявляє себе, коли відбувається карбоксилювання з утворенням малоніл-КоА у біотин залежній реакції.
В результаті цієї реакції атом вуглецю молекули СО2 стає атомом вуглецю вільної карбоксильної групи малоніл-КоА. Ацетил-КоА-карбоксилаза здатна також карбоксилювати пропіоніл-КоА з утворенням метил-малоніл-КоА, але ця реакція протікає зі значно меншою швидкістю. Активізувати процес карбоксилювання ацетил-КоА можуть цитрат, ізоцитрат, α-кетоглутарат, які являються позитивними алостеричними стимуляторами.

У реакціях полімеризації, де формальною структурною одиницею молекули знов синтезованої речовини виступає ацетат, звичайно реально бере участь малонат. Утворення малонату є ні чим іншим, як формою активації молекули ацетил-КоА, що забезпечує можливість реакції полімеризації. Вихідним для синтезу з'єднання може бути ацетил-КоА. Ріст ланцюга починається з карбоксильної групи ацетил-КоА шляхом послідовного його збільшення на два вуглецевих атоми. Кожні наступні два вуглецевих атоми (або ацетильні залишки) беруть своє походження від малоніл-КоА. У ході реакції атом вуглецю, що належить вільній карбоксильній групі, губиться у вигляді СО2. Ацетил-КоА в цій реакції виступає в електрофільній формі, а малоніл-КоА - у нуклеофільній, тобто відбувається електрофільна атака:

Таким чином, вільна карбоксильна група кожного нового залишку малоніл-КоА заміщається на карбоксил зростаючого ланцюга ацил-КоА. У результаті декарбоксилювання відбувається активація кінцевої групи внаслідок виникнення негативного заряду.
Крім реакції карбоксилювання ацетил-КоА малоніл-КоА може походити із щавлевооцтової кислоти. У результаті її декарбоксилювання утворюється малонова кислота, до якої приєднується коензим А. Щавлевооцтова кислота може виступати в реакції як донор карбоксильної групи. Каталізує реакцію фермент фосфопіруваткарбоксикіназа:

У синтезі жирних кислот і деяких поліпептидних з’єднань приймає участь ацилпереносячий білок(АПБ). У E. сoli він представляє собою білок, що складається з 77 залишків амінокислот у одному поліпептидному ланцюзі, з високим вмістом залишків кислих амінокислот (14-глутамінової і 8-аспарагінової) і з низьким вмістом лужних. Ацилпереносячі білки з різних об’єктів мають великі відмінності. Зокрема, було встановлено, що АПБ з E. сoli можуть функціонувати в системі з ферментами із вищих рослин АПБ, як і коензим А, має 4´- фосфопантотенову кислоту в якості простетичної групи. Ацетил-КоА і малоніл-КоА реагують с АПБ завдяки активності різних трансфераз. Ацетил-КоА: АПБ-трансфераза активна в присутності ацетил-КоА-похідних, переважно коротко ланцюгових.
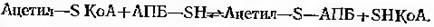
Малоніл-КоА АПБ-трансфераза специфічна для даного субстрату. Малоніл-КоА утворює проміжну сполуку у вигляді кисневого ефіру, пов’язаного з трансферазою.

Наступний етап каталізує конденсуючий фермент β-оксоацил-А-синтетаза. Фермент проводить реакції елонгації, де в якості компонентів можуть виступати ацетил-КоА, малоніл-КоА та їх коротко ланцюгові гомологи. Реакція проходить у два етапи, в результаті утворюється ацетоацетил АПБ.

Якщо походить синтез жирних кислот, то на наступному етапі працює НАД-залежний фермент.

Як вважають, участь малоніл-КоА в синтезі і виділення СО2 в результаті реакції має принципове значення для її ходу. Якби у синтезі ацетоацетил-похідного приймали участь дві молекули активного ацетату, тобто без виділення СО2, то виникла б необхідність забезпечити реакцію значною кількістю енергії, а її рівновага була б сильно здвигнута вліво. Наявність малоніл-КоА в сфері реакції веде к її здвигу в сторону синтезу і виділення тепла. Таким чином, декарбоксилування малонільного залишку забезпечує сильний термодинамічний «поштовх» у напрямку синтезу, зокрема жирних кислот. В тому випадку, коли у сфері реакції відсутній НАДН, замість жирних кислот може синтезуватися лактон, що складається з трьох ацетатних одиниць. З молекули ацетил-КоА в структуру входять С6 і метильна група при С6, інші 4 атоми вуглецю походять з малоніл-КоА.

Подальша конденсація, як бачимо, неможлива. Лактон стабілізується і втрачає зв’язок з ферментним білком. Тиольні групи АПБ відновлюються.
9.3 Циклічні структури
Загальні особливості синтезу. Ацетатна теорія синтезу отримала розповсюдження також на синтез циклічних структур, зокрема фенолів. Спершу було запропоновано, що відбувається конденсація ацетатних одиниць по типу зв’язку «голова – хвіст», або, іншими словами, шляхом взаємодії карбоксильної групи однієї молекули з метальною групою іншої. Пізніше було показано на прикладі синтезу 6-метилсаліцилової, що в реакції приймають участь одна молекула ацетил-КоА і 3 молекули малоніл-КоА, а також одна молекула НАДФН. Не дивлячись на те, що більшість ферментів, які здійснюють реакції, не описані, синтез можливо представити у вигляді ряду послідовних реакцій:

Синтетаза 6-метилсаліцилової кислоти виділена у гомогенному стані. По молекулярній масі фермент відрізняється від синтетази жирних кислот (1.3-106), але схожий з рН, інгібуванню реагентами на SH-групу та по властивості синтезувати триацетат лактон. Немає прямих доказів того, коли відбувається дегідратація. Однак, очевидно, вона змінює своє місце після відновлення при одному з атомів вуглецю. У загальній формі, без деталей і проміжних етапів, синтез фенолів і їх похідних може бути представлений у наступному вигляді: як наглядно показано на схемі, в результаті реакції може бути забезпечено достатньо велике розмаїття продуктів: а – ацильна похідна флороглюцину; в – ацильна похідна резорцину; г – можливо розглядати як похідну м-крезолу або саліцилової кислоти.
Істотною особливістю процесу синтезу циклічних структур у відмінності від жирних кислот є те, що зростаючий аліфатичний ланцюг у відомих межах підтримується без відновлення.

Характерна також можливість ідентифікації полікетометилових похідних в синтезі ароматичних продуктів. Ця обставина дає істотну можливість вважати, що має місце серія реакцій, пов’язаних з молекулою ферменту. Основними причинами, що сприяють синтезу циклічних структур, можливо, слід вважати відсутність балансу між азотом і вуглецем з перевагою останнього. Ферменти, що приймають участь у формуванні вихідних метаболітів, - ацетил-КоА і малоніл-КоА – завжди присутні у міцелію. Напрям реакцій визначається генетичними особливостями культури.
Тетрациклінові антибіотики. Як і у випадку синтезу відносно простих циклічних з’єднань, вихідним продуктом для біосинтезу антраценової структури тетрациклінових антибіотиків є ті ж КоА-похідні, що і при синтезі жирних кислот. Необхідно, однак, відмітити відмінності, які спостерігаються у ході реакції. На відміну від синтезу жирних кислот карбоксильні групи конденсуючих одиниць малоніл-КоА не відновлюються. Проміжні полікетиди, що мають тіоефірний зв’язок з ферментним білком, залишаються повністю або частково окислені і на наступних етапах конденсації. Тому тут на початкових етапах синтезу не потребується НАДФН; не проявляє активність дегідратаза, що утворює подвійний зв’язок. Її активність проявляється вже після формування циклічної структури.
Для синтезу повної молекули тетрацикліну необхідно мати лише 3 молі НАДФН, тоді як для синтезу молекули пальмінової кислоти – 14 молей НАДФН. Причому реакції з його участю можуть проходити і на циклічній структурі. Крім сказаного можна відмітити, що літогенез і біосинтез антибіотику проходять на різних етапах росту культури, тому конкуренції за вихідні продукти синтезу не відбувається.



Вихідними структурними одиницями для біосинтезу молекули тетрацикліну слугують 8 молекул малоніл-КоА і одна молекула малонаміл-КоА. Останній утворюється з аспарагіну. Можливо уявити собі два шляхи перетворення аспарагіну в малонамід. В першому з них амідна група аспарагіну зберігається в процесі утворення малонамід, у другому – амідна група малонаміду походить з аміногрупи аспарагіну. Утворений малонамід активується, в результаті чого спочатку проходить синтез ациладенілату, а потім малонаміл-КоА.
Деталі біогенезу молекули тетрацикліну (рис. 9-1) невідомі, хоча теоретичні міркування про можливі реакції привели З. Ванька і З. Гошталека до висновків, що шлях від ацетил-КоА до гіпотетичного нонакетиду вміщує у себе взаємодію приблизно 50 ферментних систем. Подальші перетворення нонакетиду у тетрациклін (або хлор тетрациклін) контролюються 11 ферментними реакціями. Загальна схема біогенезу показана на рис. 9.2.
Після синтезу нонакетиду, коли подальша конденсація, очевидно, стає неможливою, відбувається циклізація з одночасним введенням в С6- положення метальної групи. Механізм метилування розглянутий окремо. Тут слід лише відмітити, що мутанти, які синтезують 6-диметилтетрациклін, звичайно дефіцитні по параамінобензойній кислоті. Приведені у схемі проміжні продукти біосинтезу стали відомі дякуючи отриманню мутантів, у яких був блокований шлях біогенезу на різних його етапах. На схемі показано, що один з проміжних продуктів може піддаватись хлоруванню. У цьому випадку доповнюється лише одна ця реакція. Усі інші реакції на хлорованому ядрі відбуваються так як і на не хлорованому. У результаті синтезується тетрациклін або хлор тетрациклін. У обох випадках основними реакціями, що приймають участь у формуванні молекули, слід назвати наступні: циклізація, метилування у С6-положенні, гідроксилювання кільця А і С4, гідроксилювання при С12 з одночасним відновленням подвійного зв’язку (4а – 12а), амінування в С4, хлорування (у випадку синтезу хлор тетрацикліну), N-метилювання при С4, гідроксилювання при С6 і відновлення подвійного зв’язку 5а – 11а.
У багатьох реакціях синтезу, зокрема, при синтезі циклічних структур виключно важливе значення має відновлена форма НАДФ, яка утворюється в результаті ряду ферментативних реакцій. Основним постачальником НАДФН2 є реакції пентозофосфатного (гексозомонофосфатного) шляху, а у даному випадку – малатдегідрогеназа НАДФ-залежна декарбоксилююча (рис.9-3,9-4).
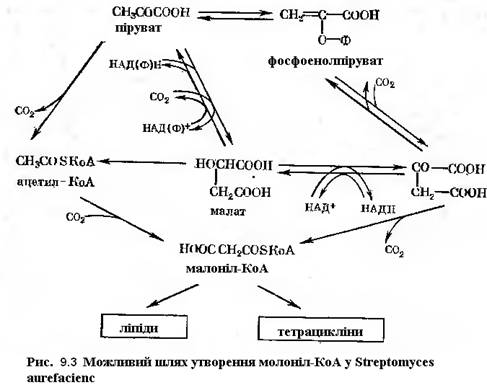
Вельми корисно при оцінці потенційної синтетазної активності культури звертати увагу на хід реакцій, в результаті яких утворюються відновлені форми НАД. У окремому випадку синтезу тетрациклінових структур основні реакції, в результаті яких утворюється НАДН2, проходять раніше ніж синтез антибіотику. Оскільки «стартові» реакції синтезу пов’язані із циклом Кребса, детальному аналізу піддаються продукти циклу, що присутні у культурі продуценту, і ферментативні реакції, що здійснюють їх перетворення. Як загальна закономірність був прийнятий факт, що чим нижче рівень ферментативних реакцій у циклі Кребса, тим вище вихід тетрацикліну. Вважають, що продуктивний штам має дефект у своєму енергообміні, дивлячись на низький рівень АТФ і низьку активність ферментів циклу. Результатом цього є посилення утилізації ацетату для синтезу вторинних метаболітів.
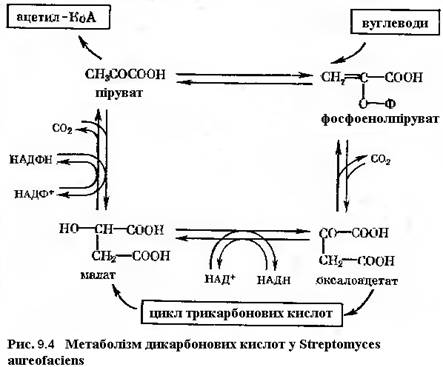
Відомо також, що при синтезі лимонної кислоти з щавелевооцтової і ацетил-КоА потрібна АТФ. Гальмує активність ферментів циклу Кребса бензилтіоцианат, який звичайно застосовують у якості компонента середовища при ферментація тетрациклінів. Внаслідок того, що біосинтез тетрацикліну проходить при пониженій активності основної енергетичної системи клітини, процес слід вести при жорсткому контролі вмісту фосфату у середовищі, хоча сам процес утворення ацетил-КоА потребує АТФ. Важливе значення у формуванні структури має щавлевооцтова кислота, при декарбоксилуванні якої утворюється малонова кислота і далі її КоА-похідна – малоніл-КоА.


Згідно рисунку 9.1, ацетил-КоА може мати походження із пірувату дякуючи активності піруватдегідрогенази. Однак у деяких продуктивних культур її активність порівняно невисока.
Вище говорилося про не збіг по часу літогенезу і синтезу тетрациклінів. Можливо, вузловим пунктом, який обумовлює перехід до вторинного метаболізму, у даному випадку – синтезу тетрацикліну, є не кінець росту культури, а сповільнення синтезу білка. Отже, синтез вторинних тетрациклінових олігокетидів може розглядатися як шунтовий метаболізм ацетату у період, коли сповільнюється синтез білку і коли ацетат перестає використовуватися в циклі Кребса в якості проміжного продукту в біосинтезі амінокислот. Таким чином, біосинтез вторинних олігокетидів розглядається як генетично фіксований шунтовий метаболізм ацетату. Поліацетильний полімер може бути кінцевим метаболітом, приймати на себе різні функціональні групи, конденсуватися з продуктами інших метаболічних шляхів, такими як цукор, ароматичні або гетероциклічні структури, що походять від шикимової кислоти.
Грізеофульвін. Молекула антибіотику гризеофульвіну синтезується з одної молекули ацетил-КоА і шести молекул малоніл-КоА(рис.9-5).
Спочатку тут синтезується полікетидна сполука, яка циклізується у похідну бензофенону. Потім проходить ряд реакцій на циклічній структурі. Спочатку метилювання, потім хлорування, повторне метилювання з утворенням гризеофенону А. останній етап – гідрування дає молекулу гризеофульвіну. Хлорування забезпечується присутнім у середовищі хлоридом колію.
9.4 Метилмалоніл-КоА, його біогенез і участь в реакціях синтезу
Іншою структурною одиницею, що приймає участь в біогенезі багатьох природних сполук, крім ацетил-КоА, є пропіонова кислота у вигляді КоА-похідної – пропіоніл-КоА.

У культурах мікроорганізмів може синтезуватися різними шляхами. Основні з них показані на рис. 9.6 як і при синтезі через ацетил-КоА, коли приймає участь малоніл-КоА, тут у реакцію вступає сполука, яка має на один вуглецевий атом більше, ніж пропіоніл-КоА – метилмалоніл-КоА. Він утворюється в результаті залежної від кобаміду ферментативної реакції мутації сукциніл-КоА, що здійснюється метилмалоніл-КоА-мутазою. При цьому проходить переміщення карбонільної тіоефірної групи до сусіднього вуглецевого атома і утворення метильної групи в наслідок переміщення атома водню. Не виключено і карбоксилювання пропіоніл-КоА дякуючи активності біотин залежної карбоксилази. Ця реакція можлива, оскільки α-С-атом пропіонової кислоти має виражений нуклеофільний характер і може заміщуватися електрофільними реагентами. Саме таким електрофільним реагентом є СО2, що вступає у вигляді карбоксибіотика.
Пропіоніл-КоА може накопичуватися в культурі в результаті функціонування системи β-окислення непарних жирних кислот, а також при ферментативній дисиміляції ізолейцину. Джерелом метилмалоніл-КоА можуть бути амінокислоти валін і ізолейцин. Очевидно, у реакціях синтезу вони мають менше значення і входять в процес лише у випадку надмірного метаболічного фонду.
Найпростішим прикладом синтезу з участю названих сполук може бути реакція між Пропіоніл-КоА і метилмалоніл-КоА, що є початковою для синтезу багатьох сполук. Тут має місце електрофільна атака:

Більш значну молекулу утворює одна молекула Пропіоніл-КоА і 6 молекул метилмалоніл-КоА при синтезі молекули макролідного антибіотику еритроміцину. Спочатку синтезується лактон еритронолід В, який далі циклізується і переходить в еритроміцин при приєднанні кладинози і диметиламіногексози – дезозаміну (рис.9-7). Як вважають, реакції проходять на одному мультиферментному комплексі, від якого молекула відщеплюється тільки після повного завершення його синтезу

Можливе одночасна участь у синтезі декількох різних структурних одиниць. Деякі мікобактерії, наприклад, утворюють С32-мікоцерозинову кислоту з 10 молекул активного ацетату і 4 молекул активного пропіонату:
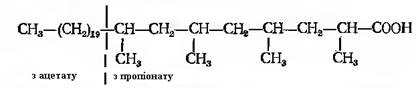
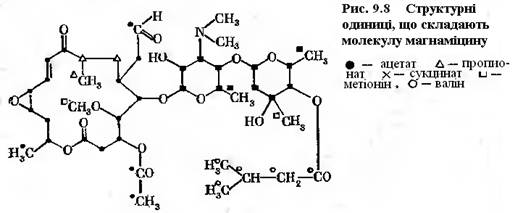
Більш значною молекулою, у формуванні якої приймають участь не лише ацетат і пропіонат, є антибіотик магнаміцин (рис.9-8)
Властивість пропіонової кислоти приймати участь у синтезі різних метаболітів може бути використана на практиці при направленому біосинтезі. При ферментації еритроміцину її можна вводити в середовище у вигляді солі або аміду. Однак практично вводять пропіловий спирт, як менш токсичну, ніж кислота, речовину. Використання пропілового спирту з ізотопними мітками по вуглецю в різних положеннях показало, що він включається в молекулу еритроноліду. Крім того, що при біосинтезі еритроміцину пропанол може включатися як структурна одиниця в молекулу, він є індуктором карбоксилази пропіоніл-КоА. У теперішній час достатньо переконливо показано, що введення пропанолу в середовище у ході ферментації може при відповідних технологічних режимах різко підвисити вихід антибіотику.
У продуцента Str. erythreus процес активації пропіонату є двоступеневою реакцією, тобто спочатку проходить кіназна реакція, як у випадку з ацетатом, а потім реакція переносу ацильної групи. Кінази відносяться до числа ферментів, що мають невисоку специфічність. Однак специфічні кінази все ж таки існують. Специфічна у відношенні пропіонату кіназа у продуцента еритроміцину. Вона вибірково фосфорилює пропіонат, тому відбір високо продуктивного штаму еритроміцину може проходити по критерію специфічності кінази до пропіонату. У декількох відомих штамів-продуцентів еритроміцину висока специфічна кіназна активність відносно пропіонату корелює з високою продуктивністю мікроорганізму.
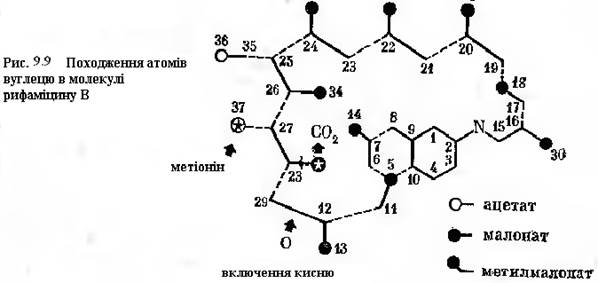
Прикладом достатньо складної структури, сформованої з ацетату, малонату, метилмалонату, може служити антибіотик рифаміцин. Розподіл названих сполук по вуглецевій структурі антибіотику показано на рис.9-9. Вважають, що полікетидний ланцюг утворюється шляхом лінійної конденсації 8 метилмалонатних одиниць і 2 малонатних одиниць з наступною еритрифікацією гідроксилу ацетатом С-25-атома. У формуванні хромофорної частини приймає участь глюкоза і гліцерат. Реальними метаболічними попередниками є проміжні метаболіти загального шляху біосинтезу ароматичних сполук, однак не шики мова кислота. Тільки 7 вуглецевих атомів у рафіміцині походять не з пропіонату і ацетату.
9.5 Мевалонова кислота, її біогенез і участь в реакціях синтезу
Каротиноїди. Серед продуктів обміну речовин мікроорганізмів зустрічаються такі, структура яких формально відповідає повторювальним ізопреновим одиницям. Однією із важливих подібних структур є каротиноїди. За класичним визначенням каротиноїди це жовті або червоні пігменти аліфатичної чи аліциклічної будови, які складаються з ізопренових залишків. Останні сполучені таким чином, що дві найближчі до центру молекули метильні групи знаходяться в положенні 1:6, тоді як усі інші бокові метильні групи розташовані в положеннях 1:5. Ряд подвійних зв’язків складає хромофорну систему каротиноїдів (рис. 9.10).

Найбільше значення має і представляє практичне значення β-каротин. Молекула його має 2 β-іонові групи.

Вивчення закономірностей біосинтезу каротиноїдів проводилось переважно з культурами Blakeslea trispora і Phycomyces blakesleeanus і деякими дріжджами. Найбільш інтенсивно синтез β-каротину проходить після того, як ріст кільтури практично припинений (рис. 9.11). Каротиноїди мають внутрішньоклітинну локалізацію. У названих вище культур грибів вони асоційовані з ліпідами, рідше – з білками. Каротиноїди можуть бути включені в структуру цитоплазматичних мембран, розташовуючись у жирових гранулах. Внаслідок гарної розчинності в жирах каротиноїди можуть бути в комплексі з вільними ліпідами клітини.

Біосинтез каротиноїдів складається з наступних етапів: 1) синтез первинного С5-попередника; 2) утворення безкольорових С40-з’єднань з С5-попередника; 3) формування молекул різних каротиноїдів шляхом дегідрування безкольорового попередника; 4) циклізація.
Перший етап можна розглядати як приватний випадок синтезу через ацетил-КоА, так як саме цей продукт приймає участь у перших трьох реакціях. Продуктом цих реакцій є 3-окси-3-метилглутарил-КоА, який в результаті НАД-залежного ензиматичного відновлення перетворюється в мевалонову кислоту. Проміжним продуктом, який не виступає у вільній формі, є мевальдинова кислота. Мевалонова кислота розглядається як прямий і специфічний попередник ізопренових похідних. Слід відмітити, що мевалонова кислота може бути отримана при культивуванні одного з мутантів штаму Endomyces fibuliger.
Далі в ланцюгу біогенезу каротиноїдів мевалонова кислота піддається фосфорилюванню АТФ, що протікає у 3 етапи. На першому з них утворюється 5-фосфомевалонова кислота, далі – 5-пірофосфомевалонова. Третя реакція фосфорилюванню супроводжується декарбоксилуванням, в результаті якого утворюється ізопентенілпірофосфат. Останній може піддаватись ізомеризації в 3,3-диметилалілпірофосфат. Біосинтез безкольорових С40-з’єднань з С5-попередника починається з утворення геранілпірофосфату, що проходить при нуклеофільній атаці диметилалілпірофосфату ізопентенілпірофосфатом.
Подальші трансферазні реакції приводять до утворення фернезилпірофосфату, з’єднання, що містить 15 вуглецевих атомів і геранілгеранілпірофосфату з 20 вуглецевими атомами (рис.9-12). Димерізація останнього приводить до отримання першого безкольорового С40-попередника β-каротину – фітоїну.
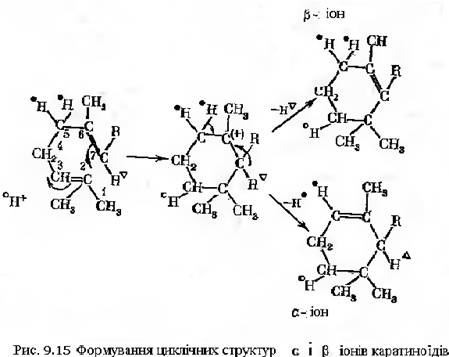
В результаті послідовно протікаючих реакцій дегідрування фітону утворюється фітофлуїн, ζ-каротин, нейроспорин, лікопін. В ході реакцій фітон ступінчато губить 8 атомів водню (рис.9-13). Згідно схеми лікопін виступає в якості кінцевого продукту, однак в деяких організмах можуть функціонувати ферментні системи, що здійснюють його перетворення в β-каротин. Кінцевим етапом біосинтезу каротиноїдів є циклізація з утворенням α- і β-іонових кілець (рис.9-14). Процес циклізації починається з приєднання протону до третього вуглецевого атому, відновлення подвійного зв’язку між 2 і 3 вуглецевими атомами з одночасним утворенням зв’язку між 2 і 3 атомами. Остання реакція веде до відновлення подвійного зв’язку між 6 і 7 атомами. Далі, у випадку утворення β-іонової структури атом водню відщеплюється від 5-го вуглецевого атома. Відповідно між 6 і 7, 5 і 6 атомами вуглецю утворюються подвійні зв’язки.


Великий вклад у встановлення проміжних продуктів біосинтезу каротиноїдів внесли досліди по вивченню дії інгібіторів і стимуляторів. Найбільш відомою сполукою, при додаванні якої в середовища відбувається пригнічення синтезу каротиноїдів, є дифеніламін:

Останній перешкоджає процесу дегідрування, викликаючи накопичення більш насичених сполук, в першу чергу фітоїну і фітофлуїну.
Стимулюючий ефект на біосинтез каротиноїдів, в першу чергу β-каротину, здійснює β-іон. Так як циклічна структура молекули β-каротину подібна за будовою з β-іоном, передбачалось, що його стимулюючий ефект можна пояснити безпосереднім включенням в молекулу каротиноїду. Однак численні іспити, що проводились з 14С-β-іоном, показали, що останній не є метаболічним попередником β каротину, оскільки включення мітки від іону в структуру β-іону каротиноїдів не перевищує 1-3%. В культурі Phycomyces blakesleanus β-іон виконує функцію неспецифічного регулятора ензиматичної реакції. β-іон знімає інгібування кінцевим продуктом на рівні реакції 5-фосфомевалонова кислота – диметилалілпірофосфат.
Іспити з Blakeslea trispora призвели до доказу збільшення синтезу каротину, білку і РНК при додаванні β-іону до зростаючої культури, при чому було зроблено висновок, що стимуляція процесу здійснюється на рівні трансляції. Як бачимо, має велике значення, в якому вигляді вносити β-іон в середовище. Так, на відміну від Phycomyces blakesleanus в культурі Blakeslea trispora стимулююча дію β-іона проявляється тільки в присутності масел, причому склад ліпідів виявляє суттєвий вплив на утворення β-каротину (табл.9-1):

Речовинами, що можуть стимулювати синтез біотину β-каротину, є триспорові кислоти. Відомі триспорові кислоти А, Б,С, що входять до складу фактора β. По хімічній природі вони представляють собою окислені ненасичені похідні 1,1´,3-триметил-2(3метил)-циклогексана. Звичайно переважає триспорова кислота С, що складає близько 90% їх суми.

Триспорові кислоти можна розглядати як статевий гормон або сексуальний фактор гетерогалічних грибів, що мають штами (-) і (+). Такими гетероталічними грибами є Blakeslea trispora. Наявність в клітинах фактору β веде до утворення зиготи. Для виділення фактора β використовують сумісно зростаючі культури штамів (+) і (-) гетероталічних грибів. Аналіз культуральної рідини окремо зростаючих штамів показав, що в цих умовах фактор β не утворюється. Він є не тільки статевим гормоном, що регулює одну з стадій репродукції, але і ініціатором утворення каротину у окремо зростаючих штамів деяких нижчих грибів. В молекулу каротину фактор β не включається і в досить незначних кількостях витрачається в процесі культивування гриба і біосинтезу β-каротину.
Вважають, що триспорові кислоти стимулюють синтез ферментів, що діють на етапі від ацетил-КоА до β-каротину. Механізм дії триспорових кислот полягає у тому, що вони є дипресором гену, що регулює синтез специфічних ферментів, які приймають участь в синтезі β-каротину.
Найбільш помітний ефект триспорові кислоти виявляють при введенні в культуру (-) штаму Blakeslea trispora. Тут ефект збільшення синтезу β-каротину може досягати 25-30 разів. На синтез β-каротину (+) штамом ефект не такий значний, не перевищує 1,5-2 різів. Позитивний ефект найбільш помітний при внесенні триспорової кислоти В; він залежить від часу введення в середовище і концентрації триспорових кислот, яка для досягнення позитивного ефекту повинна бути достатньо високою, близько 250-500 мкг на 1мл середовища.
Гібереліни. До числа продуктів, що синтезуються через мевалонову кислоту, відносяться гібереліни. вони представляють собою групу фізіологічно активних сполук, регуляторів росту рослин, що мають гормональну природу. У теперішній час описано близько 40 різних сполук, об’єднаних загальною назвою гібереліни.

Промислове отримання гіберелінів здійснюється при культивуванні грибів, переважно різних штамів Gibberella fujikuri. По хімічній природі всі вони є тетрациклічними карбоновими кислотами, що відносяться до терпеноїдів (рис.9.16).

Гібереліни позначають символом А з цифрою справа знизу, порядковим номером, який привласнюється кожному новому гібереліну по мірі виділення і індинтифікації. В залежності від числа вуглецевих атомів гібереліни ділять на дві групи. До одної з них відносять сполуки, що мають 20 атомів вуглецю. Це істинні де терпени, число атомів вуглецю в їх молекулі кратне 5. До іншої групи належать С20 вуглецеві сполуки. Основні відмінності між гіберелінами спостерігаються у відношенні замісників у кільці А С19-гібереліни містять лактонну структуру.
Найбільш розповсюдженим і одним з найбільш фізіологічно активних гіберелінів є речовина А3, яке отримало назву гіберелінової кислоти. Початкові етапи синтезу її молекули проходять по шляху, подібному до шляху каротиноїдів. Загальний кінцевий продукт у обох процесах синтезу – герацилгеранілпірофосфат.
Етапи синтезу, що ведуть до гіберелінів, менш вивчені, ніж попередні. Можливо, що не всі проміжні сполуки повністю ідентифіковані. Тим не менш, загальний хід процесу відомий. Протиціанізована циклізація герацилгеранілфосфату приводить до утворення біциклічної структури. Депротеїнізація останнього дає лабадієн, який далі циклізується до пімарадієну. Останній слугує вихідним продуктом синтезу (-)-каурена з нього при дії оксигеназ зі змішаною функцією утворюється спочатку кауренол, потім каурекаль, кауренова кислота, 7-оксикауренова кислота (рис.9.17)