2018-01-21
2018-01-21 11778
11778Представление о катализе:
Катализ — избирательное ускорение одного из возможных термодинамически разрешенных направлений химической реакции под действием катализатора(ов), который многократно вступает в промежуточное химическое взаимодействие с участниками реакции и восстанавливает свой химический состав после каждого цикла промежуточных химических взаимодействий.
Катализатор изменяет механизм реакции на энергетически более выгодный, то есть снижает энергию активации. Катализатор образует с молекулой одного из реагентов промежуточное соединение, в котором ослаблены химические связи. Это облегчает его реакцию со вторым реагентом. Важно отметить, что катализаторы ускоряют обратимые реакции, как в прямом, так и в обратном направлениях.
Катализ гомогенный и гетерогенный:
При гомогенном катализе действие катализатора связано с тем, что он вступает во взаимодействие с реагирующими веществами с образованием промежуточных соединений, это приводит к снижению энергии активации.
При гетерогенном катализе ускорение процесса обычно происходит на поверхности твердого тела — катализатора, поэтому активность катализатора зависит от величины и свойств его поверхности. На практике катализатор обычно наносят на твердый пористый носитель.
Механизм гетерогенного катализа сложнее, чем у гомогенного. Механизм гетерогенного катализа включает пять стадий, причем все они обратимы.
1. Диффузия реагирующих веществ к поверхности твердого вещества
2. Физическая адсорбция на активных центрах поверхности твердого вещества реагирующих молекул и затем хемосорбция их
3. Химическая реакция между реагирующими молекулами
4. Десорбция продуктов с поверхности катализатора
5. Диффузия продукта с поверхности катализатора в общий поток
Автокатализ:
Автокатализ — катализ химической реакции одним из её продуктов или исходных веществ. Одним из наиболее широко известных примеров автокатализа является окисление щавелевой кислоты перманганатом калия:
2MnO4− + 5C2O42− + 16H+ = 2Mn2+ + 10CO2 + 8H2O
Катализатором этой реакции являются ионы Mn2+. При комнатной температуре эта реакция вначале протекает медленно, но по мере накопления в растворе продукта-катализатора, она ускоряется.
Применения катализа:
Биологические катализаторы, называемые ферментами, участвуют в регуляции биохимических процессов. Без катализаторов не могли бы протекать многие промышленные процессы. Важнейшее свойство катализаторов - селективность, т.е. способность увеличивать скорость лишь определенных химических реакций из многих возможных. Это позволяет осуществлять реакции, протекающие в обычных условиях слишком медленно, чтобы им можно было найти практическое применение, и обеспечивает образование нужных продуктов. Применение катализаторов способствовало бурному развитию химической промышленности. Они широко используются при переработке нефти, получении различных продуктов, создании новых материалов (например, пластмасс), нередко более дешевых, чем применявшиеся прежде. Примерно 90% объема современного химического производства основано на каталитических процессах. Особую роль играют каталитические процессы в охране окружающей среды.
Кислородные и водородные соединения. Оксиды и их гидратные формы. Кислоты и основания. Соли, классификация. Генетическая связь между классами неорганических соединений, способы получения, свойства.
Кислородные и водородные соединения:
С кислородом неметаллы образуют кислотные оксиды. В одних оксидах они проявляют максимальную степень окисления, равную номеру группы (например, N2O5), а других – более низкую (например, SO2, N2O3). Кислотным оксидам соответствуют кислоты, причем из двух кислородных кислот одного неметалла сильнее та, в которой он проявляет более высокую степень окисления. Например, азотная кислота HNO3 сильнее азотистой HNO2, а серная кислота H2SO4 сильнее сернистой H2SO3.
Характеристики кислородных соединений неметалов:
1. Свойства высших оксидов (т.е. оксидов, в состав которых входит элемент данной группы с высшей степенью окисления) в периодах слева направо постепенно изменяются от основных к кислотным.
2. В группах сверху вниз кислотные свойства высших оксидов постепенно ослабевают. Об этом можно судить по свойствам кислот, соответствующих этим оксидам.
3. Возрастание кислотных свойств высших оксидов соответствующих элементов в периодах слева направо объясняется постепенным возрастанием положительного заряда ионов этих элементов.
4. В главных подгруппах периодической системы химических элементов в направлении сверху вниз кислотные свойства высших оксидов неметаллов уменьшаются.
С металлами водород образует (за некоторым исключением) нелетучие соединения, которые являются твердыми веществами немолекулярного строения. Поэтому их температуры плавления сравнительно высоки.
С неметаллами водород образует летучие соединения молекулярного строения. В обычных условиях это газы или летучие жидкости.
В периодах слева направо кислотные свойства летучих водородных соединений неметаллов в водных растворах усиливается. Это объясняется тем, что ионы кислорода имеют свободные электронные пары, а ионы водорода – свободную орбиталь
Оксиды и их гидратные формы:
Оксиды подразделяются на солеобразующие и несолеобразующие.
Солеобразующими называют такие оксиды, которые в результате химических реакций способны образовывать соли.
Несолеобразующие оксиды такой способностью не обладают. Примером несолеобразующих оксидов могут служить следующие вещества: CO, N2O, NO.
Солеобразующие оксиды, в свою очередь подразделяются на основные, кислотные и амфотерные.
Основными оксидами называются такие оксиды, которым в качестве гидратов (продуктов присоединения воды) соответствуют основания.
Кислотными оксидами называются такие оксиды, которым в качестве гидратов соответствуют кислоты. Кислотные оксиды называют также ангидридами кислот.
Амфотерные оксиды представляют собой оксиды, которые в зависимости от условий проявляют свойства как основных (в кислой среде), так и кислотных (в щелочной среде) оксидов.
Кислоты и основания:
Кислоты — сложные вещества, в состав которых обычно входят атомы водорода, способные замещаться на атомы металлов, икислотный остаток. Водные растворы кислот имеют кислый вкус, обладают раздражающим действием, способны менять окраскуиндикаторов, отличаются рядом общих химических свойств.
Классификация кислот:
1. По содержанию кислорода
· бескислородные (HCl, H2S);
· кислородосодержащие (HNO3,H2SO4).
2. По основности — количество кислых атомов водорода
· Одноосновные (HNO3);
· Двухосновные (H2SeO4, двухосновные предельные карбоновые кислоты);
· Трёхосновные (H3PO4, H3BO3).
· Полиосновные (практически не встречаются).
3. По силе
· Сильные — диссоциируют практически полностью, константы диссоциации больше 1·10−3 (HNO3);
· Слабые — константа диссоциации меньше 1·10−3 (уксусная кислота Kд= 1,7·10−5).
4. По устойчивости
· Устойчивые (H2SO4);
· Неустойчивые (H2CO3).
5. По принадлежности к классам химических соединений
· Неорганические (HBr);
· Органические (HCOOH,CH3COOH);
6. По летучести
· Летучие (H2S, HCl);
· Нелетучие (H2SO4);
7. По растворимости в воде
· Растворимые (H2SO4);
· Нерастворимые (H2SiO3);
Основания — класс химических соединений.
· Основания (основные гидроксиды) — сложные вещества, которые состоят из атомов металла или иона аммония и гидроксогруппы (-OH). В водном растворедиссоциируют с образованием катионов и анионов ОН−. Название основания обычно состоит из двух слов: «гидроксид металла/аммония». Хорошо растворимые в воде основания называются щелочами.
· Согласно протонной теории кислот и оснований, основания — один из основных классов химических соединений, вещества, молекулы которых являютсяакцепторами протонов.
· В органической химии по традиции основаниями называют также вещества, способные давать аддукты («соли») с сильными кислотами, например, многиеалкалоиды описывают как в форме «алкалоид-основание», так и в виде «солей алкалоидов».
Классификация оснований:
1. По растворимости в воде.
· Растворимые основания (щёлочи): гидроксид лития LiOH, гидроксид натрия NaOH, гидроксид калия KOH, гидроксид барияBa(OH)2, гидроксид стронция Sr(OH)2, гидроксид цезия CsOH, гидроксид рубидия RbOH.
· Практически нерастворимые основания: Mg(OH)2, Ca(OH)2, Zn(OH)2, Cu(OH)2, Al(OH)3, Fe(OH)3, Be(OH)2.
· Другие основания: NH3·H2O
2. По количеству гидроксильных групп в молекуле.
· Однокислотные (гидроксид натрия NaOH)
· Двукислотные (гидроксид меди(II) Cu(OH)2)
· Трехкислотные (гидроксид железа(III) Fe(OH)3)
3. По летучести.
· Летучие: NH3, CH3-NH2
· Нелетучие: щёлочи, нерастворимые основания.
4. По стабильности.
· Стабильные: гидроксид натрия NaOH, гидроксид бария Ba(OH)2
· Нестабильные: гидроксид аммония NH3·H2O (гидрат аммиака).
5. По степени электролитической диссоциации.
· Сильные (α > 30 %): щёлочи.
· Слабые (α < 3 %): нерастворимые основания.
6. По наличию кислорода.
· Кислородсодержащие: гидроксид калия KOH, гидроксид стронция Sr(OH)2
· Бескислородные: аммиак NH3, амины.
7. По типу соединения:
· Неорганические основания: содержат одну или несколько групп -OH.
· Органические основания: органические соединения, являющиеся акцепторами протонов: амины, амидины и другие соединения.
Соли, классификация:
Соли — класс химических соединений, состоящих из катионов и анионов.
В роли катионов в солях могут выступать катионы металлов, ониевые катионы (катионов аммония 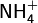 , фосфония
, фосфония 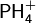 , гидроксония
, гидроксония 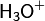 и их органические производные), комплексные катионы и т.д., в качестве анионов — анионы кислотного остатка различных кислот Бренстеда - как неорганических, так и органических, включая карбанионы, комплексные анионы и т.п.
и их органические производные), комплексные катионы и т.д., в качестве анионов — анионы кислотного остатка различных кислот Бренстеда - как неорганических, так и органических, включая карбанионы, комплексные анионы и т.п.
· Средние (нормальные) соли — все атомы водорода в молекулах кислоты замещены на атомы металла. Пример: 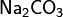 ,
, 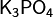 .
.
· Кислые соли — атомы водорода в кислоте замещены атомами металла частично. Они получаются при нейтрализации основания избытком кислоты. Пример: 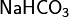 ,
, 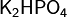 .
.
· Основные соли — гидроксогруппы основания (OH−) частично замещены кислотными остатками. Пример: 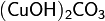 .
.
· Двойные соли — в их составе присутствует два различных катиона, получаются кристаллизацией из смешанного раствора солей с разными катионами, но одинаковыми анионами. Пример: 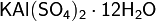 .
.
· Смешанные соли — в их составе присутствует два различных аниона. Пример: 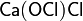 .
.
· Гидратные соли (кристаллогидраты) — в их состав входят молекулы кристаллизационной воды. Пример: 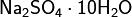 .
.
· Комплексные соли — в их состав входит комплексный катион или комплексный анион. Пример: 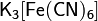 ,
, 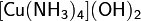 .
.
Особую группу составляют соли органических кислот, свойства которых значительно отличаются от свойств минеральных солей. Некоторые из них можно отнести к особенному классу органических солей, так называемых ионных жидкостей или по-другому «жидких солей», органических солей с температурой плавления ниже 100 °C.
Генетическая связь между классами неорганических соединений, способы получения, свойства:
Генетическая связь – это связь между веществами, которые относятся к разным классам.
Основные признаки генетических рядов:
1. Все вещества одного ряда должны быть образованы одним химическим элементом.
2. Вещества, образованные одним и тем же элементом, должны принадлежать к различным классам химических веществ.
3. Вещества, образующие генетический ряд элемента, должны быть связаны между собой взаимопревращениями.
Таким образом, генетическим называют ряд веществ, которые представляют разные классы неорганических соединений, являются соединениями одного и того же химического элемента, связаны взаимопревращениями и отражают общность происхождения этих веществ.
1. Генетический ряд металлов, гидроксиды которых являются основаниями (щелочами):
металл → основный оксид → основание (щелочь) → соль.
2. Генетический ряд металлов, которые образуют амфотерные гидроксиды:
металл → амфотерный оксид → (соль) → амфотерный гидроксид → соль
↓
Кислота
3. Генетический ряд неметаллов (неметаллы образуют только кислотные оксиды):
неметалл → кислотный оксид → кислота → соль
Коллигативные свойства растворов. Законы Рауля. Конденсированное состояние вещества. Фазовые диаграммы. Диаграмма состояния воды. Эбуллиоскопическая и криоскопическая константы, физический смысл и размерность. Осмос, движущая сила. Осмотическое давление. Правило Вант-Гоффа. Обратный осмос. Методы определения молекулярных масс.
Коллигативные свойства растворов:
Коллигативные свойства растворов — это те свойства, которые при данных условиях оказываются равными и независимыми от химической природы растворённого вещества; свойства растворов, которые зависят лишь от количества кинетических единиц и от их теплового движения.
Изменения термодинамических свойств растворов относительно свойств растворителя:
· понижение давления пара,
· повышение температуры кипения,
· понижение температуры замерзания,
Законы Рауля:
Первый закон Рауля связывает давление насыщенного пара над раствором с его составом; он формулируется следующим образом:
· Парциальное давление насыщенного пара компонента раствора прямо пропорционально его мольной доле в растворе, причём коэффициент пропорциональности равен давлению насыщенного пара над чистым компонентом.
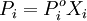
Для бинарного раствора, состоящего из компонентов А и В (компонент А считаем растворителем) удобнее использовать другую формулировку:
· Относительное понижение парциального давления пара растворителя над раствором не зависит от природы растворённого вещества и равно его мольной доле в растворе.
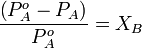
Растворы, для которых выполняется закон Рауля, называются идеальными. Идеальными при любых концентрациях являются растворы, компоненты которых очень близки по физическим и химическим свойствам (оптические изомеры, гомологи и т. п.), и образование которых не сопровождается изменением объёма и выделением либо поглощением теплоты. В этом случае силымежмолекулярного взаимодействия между однородными и разнородными частицами примерно одинаковы, и образование раствора обусловлено лишь энтропийным фактором.
Тот факт, что давление паров над раствором отличается от давления паров над чистым растворителем, существенно влияет на процессы кристаллизации и кипения. Из первого закона Рауля выводятся два следствия, касающиеся понижения температуры замерзания и повышения температуры кипения растворов, которые в объединённом виде известны как второй закон Рауля.
Разность между температурой кристаллизации растворителя T°fr и температурой начала кристаллизации раствора Tfr есть понижение температуры кристаллизации.
· Понижение температуры кристаллизации бесконечно разбавленных растворов не зависит от природы растворённого вещества и прямо пропорционально моляльной концентрации раствора. 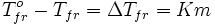
Поскольку по мере кристаллизации растворителя из раствора концентрация последнего возрастает, растворы не имеют определённой температуры замерзания и кристаллизуются в некотором интервале температур.
Жидкость кипит при той температуре, при которой общее давление насыщенного пара становится равным внешнему давлению. Если растворённое вещество нелетучее (то есть давлением его насыщенных паров над раствором можно пренебречь), то общее давление насыщенного пара над раствором равно парциальному давлению паров растворителя. В этом случае давление насыщенных паров над раствором при любой температуре будет меньше, чем над чистым растворителем, и равенство его внешнему давлению будет достигаться при более высокой температуре. Таким образом, температура кипения раствора нелетучего вещества Tb всегда выше, чем температура кипения чистого растворителя при том же давлении T°b.
· Повышение температуры кипения бесконечно разбавленных растворов нелетучих веществ не зависит от природы растворённого вещества и прямо пропорционально моляльной концентрации раствора 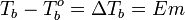
Конденсированное состояние вещества:
Конденсированное состояние вещества, твёрдое и жидкое состояния вещества. В отличие от газообразного состояния, у вещества в конденсированном состоянии существует упорядоченность в расположении частиц (ионов, атомов, молекул). Кристаллические твёрдые тела обладают высокой степенью упорядоченности — дальним порядком в расположении частиц. Частицы жидкостей и аморфных твёрдых тел располагаются более хаотично, для них характерен ближний порядок.Свойства веществ в конденсированном состоянии определяются их структурой и взаимодействием частиц.
Фазовые диаграммы:
Фазовая диаграмма - графическое изображение всех возможных состояний термодинамической системы в пространстве основных параметров состояния температуры Т, давления р и состава х (обычно выражаемого молярными или массовыми долями компонентов). Для сложных систем, состоящих из многих фаз и компонентов, построение диаграммы состояния является единственным методом, позволяющим на практике установить, сколько фаз и какие конкретно фазы образуют систему при данных значениях параметров состояния. Каждое реально существующее состояние системы на диаграмме состояния изображается так называемой фигуративной точкой; областям существования одной фазы отвечают участки пространства (на трехмерных диаграммах состояния) или плоскости (на двухмерных диаграммах состояния), условиям сосуществования фаз - соответствуют поверхности или линии; изменение фазового состояния системы рассматривается как движение фигуративной точки на диаграмме состояния.
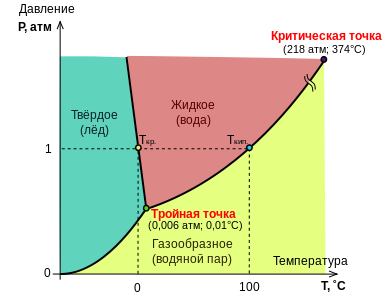
Диаграмма состояния воды:
Эбуллиоскопическая и криоскопическая константы, физический смысл и размерность:
Коэффициенты пропорциональности: К и Е — соответственно криоскопическая и эбулиоскопическая постоянные растворителя, имеющие физический смысл понижения температуры кристаллизации и повышения температуры кипения раствора с концентрацией 1 моль/кг. Для воды они равны 1.86 и 0.52 K·моль−1·кг соответственно. Поскольку одномоляльный раствор не является бесконечно разбавленным, второй закон Рауля для него в общем случае не выполняется, и величины этих констант получают экстраполяцией зависимости из области малых концентраций до m = 1 моль/кг.
Для водных растворов в уравнениях второго закона Рауля моляльную концентрацию иногда заменяют молярной. В общем случае такая замена неправомерна, и для растворов, плотность которых отличается от 1 г/см³, может привести к существенным ошибкам.
Второй закон Рауля даёт возможность экспериментально определять молекулярные массы соединений, неспособных к диссоциации в данном растворителе; его можно использовать также для определения степени диссоциации электролитов.
Осмос, движущая сила:
Осмос — процесс односторонней диффузии через полупроницаемую мембрану молекул растворителя в сторону большей концентрации растворённого вещества (меньшей концентрации растворителя).
Более широкое толкование явления осмоса основано на применении Принципа Ле Шателье — Брауна: если на систему, находящуюся в устойчивом равновесии, воздействовать извне, изменяя какое-либо из условий равновесия (температура, давление, концентрация, внешнее электромагнитное поле), то в системе усиливаются процессы, направленные на компенсацию внешнего воздействия.
Явление осмоса наблюдается в тех средах, где подвижность растворителя больше подвижности растворённых веществ.
Осмотическое давление возникает соответственно Принципу Ле Шателье из-за того, что система пытается выровнять концентрацию раствора в обоих средах, разделенных мембраной, и описывается вторым законом термодинамики. Оно равно избыточному внешнему давлению, которое следует приложить со стороны раствора, чтобы прекратить процесс, то есть создать условия осмотического равновесия. Превышение избыточного давления над осмотическим может привести к обращению осмоса — обратной диффузии растворителя.
Правило Вант-Гоффа:
Законы Рауля не выполняются для растворов (даже бесконечно разбавленных), которые проводят электрический ток — растворов электролитов. Для учёта этих отклонений Вант-Гофф внёс в приведённые выше уравнения поправку — изотонический коэффициент i, неявно учитывающий диссоциацию молекул растворённого вещества:
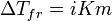 ;
; 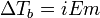
Неподчинение растворов электролитов законам Рауля и принципу Вант-Гоффа послужили отправной точкой для создания С. А. Аррениусом теории электролитической диссоциации.
Обратный осмос:
Обратный осмос — процесс, в котором с помощью давления принуждают растворитель (обычно вода) проходить черезполупроницаемую мембрану из более концентрированного в менее концентрированный раствор, то есть в обратном дляосмоса направлении. При этом мембрана пропускает растворитель, но не пропускает некоторые растворённые в нём вещества. Обратный осмос используют с 1970-х при очистке воды, получении питьевой воды из морской воды, получения особо чистой воды для медицины, промышленности и других нужд. С помощью обратного осмоса можно производить концентраты соков без нагрева.
Методы определения молекулярных масс:
На основании закона Авогадро находят молекулярную массу газообразных веществ.
Молекулярную массу трудноиспаряющихся веществ определяют, исследуя свойства их растворов.
Метод Бекмана. В этом методе измеряют температуру замерзания известного количества растворителя (w1), затем добавляют в него заданное количество растворенного вещества (w2) и измеряют понижение температуры замерзания раствора с помощью термометра Бекмана. Этот термометр регистрирует не саму температуру, а разность температур, но с точностью 0,001° С. При измерениях могут возникать ошибки, связанные с переохлаждением раствора. Для их устранения применяют более совершенные модификации прибора. Для приближенной оценки мол.массы существует более простой метод Раста, где в качестве растворителя используют камфору, температура замерзания которой при растворении в ней различных веществ понижается очень сильно и может быть измерена обычным термометром.
Методы Ландсбергера и Котрелла. Бекман сконструировал также прибор для измерения повышения температуры кипения растворов, но здесь возникают те же проблемы, что и при измерении точки замерзания, а именно связанные с перегревом раствора. Для приближенных оценок используется метод Ландсбергера, в котором жидкость нагревают до температуры кипения, пропуская через нее пар. Для точных определений предпочтительна методика Котрелла. В ней термометр не погружают в жидкость, а помещают над ее поверхностью, так что пузырьки пара, поднимающиеся вверх, увлекают за собой жидкость, и она омывает термометр. Это позволяет избежать ошибок, связанных с перегревом.
Еще один метод определения молекулярной массы растворенных веществ основан на измерении осмотического давления. Для разбавленного раствора, содержащего известное количество w граммов растворенного вещества с мол. массой М в объеме растворителя V, осмотическое давление Р при температуре Т равно P = wRT/MV. Если Р выражено в атм., а V - в см3 или мл, то константа R = 82,06. Измерить осмотическое давление растворов для обычных веществ довольно трудно. Однако этот метод оказался весьма полезным для определения мол.масс высокомолекулярных соединений, поскольку создаваемое ими осмотическое давление достаточно велико и можно получить точные данные на относительно простой аппаратуре. Высокомолекулярные соединения имеют большое практическое значение, поэтому методы определения их молекулярных масс совершенствуются. Можно упомянуть методы, основанные на измерении вязкости и рассеяния света, а также ультрацентрифугирование. Последний применяется наиболее широко для определения мол.масс биополимеров (нуклеиновых кислот и белков).
Масс-спектрометрический метод. Этим принципиально иным, чем все рассмотренные выше, методом определяют массу разных видов молекул или разных изотопов, находящихся в исследуемом объеме. Особую ценность он представляет для изотопного анализа. Допустим, требуется определить, содержится ли в образце метана СН4 изотоп 13С помимо обычного изотопа 12С. У обычного метана молекулярная масса равна 16, а у его изотопического варианта 17. В масс-спектре ему соответствует отдельная линия, по положению которой можно точно определить молекулярную массу.
Растворы электролитов. Электролитическая диссоциация. Изотонический коэффициент. Физико-химическая теория растворов. Диэлектрическая проницаемость растворителя. Степень электролитической диссоциации, связь с изотоническим коэффициентом. Классификация электролитов по силе. Кажущаяся степень диссоциации. Активность (Льюис). Коэффициент активности. Понятие о теории Дебая-Хюккеля. Ионная сила. Солевой эффект.
Растворы электролитов содержат в заметных концентрациях ионы-катионы и анионы, образующиеся в результате электролитической диссоциации молекул растворенного вещества. Растворитель (чистый или смешанный) обычно в сколько-нибудь значительной степени не диссоциирован. Р. э. обладают способностью проводить электрический ток и относятся к проводникам второго рода. Благодаря увеличению общего числа частиц коллигативные свойства бесконечно разбавленных Р. э. (т. е. свойства, зависящие только от концентрации растворенного вещества, но не от его природы) существенно отличаются от тех же свойств растворов неэлектролитов. Этим, в частности, объясняется увеличение осмотического давления в сравнении со значением, предсказываемым законом Вант-Гоффа понижение давления пара растворителя над раствором в сравнении с предсказываемым Рауля законом и др. Наличием ионов обусловлены также классификация Р. э., особенности теоретических подходов в сравнении с другими классами растворов. Наиболее изучены водные Р. э., играющие важную роль во многих биологических, геологических и технических процессах. Неводные Р. э. служат средой для проведения синтеза и электрохимических процессов, используются в современных технологиях (создание новых химических источников тока, солнечных батарей, процессы разделения веществ и др.).
Электролитическая диссоциация:
Электролитическая диссоциация — процесс распада электролита на ионы при его растворении или плавлении.
Диссоциация на ионы в растворах происходит вследствие взаимодействия растворённого вещества с растворителем; по данным спектроскопических методов, это взаимодействие носит в значительной мере химический характер. Наряду с сольватирующей способностью молекул растворителя определённую роль в электролитической диссоциации играет также макроскопическое свойство растворителя — его диэлектрическая проницаемость.
Классическая теория электролитической диссоциации была создана С. Аррениусом и В. Оствальдом в 1887 году. Аррениус придерживался физической теории растворов, не учитывал взаимодействие электролита с водой и считал, что в растворах находятся свободные ионы. Русские химики И. А. Каблуков и В. А. Кистяковскийприменили для объяснения электролитической диссоциации химическую теорию растворов Д. И. Менделеева и доказали, что при растворении электролита происходит его химическое взаимодействие с водой, в результате которого электролит диссоциирует на ионы.
Классическая теория электролитической диссоциации основана на предположении о неполной диссоциации растворённого вещества, характеризуемой степенью диссоциации α, т. е. долей распавшихся молекул электролита. Динамическое равновесие между недиссоциированными молекулами и ионами описывается законом действующих масс. Например, электролитическая диссоциация бинарного электролита KA выражается уравнением типа: 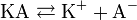
Константа диссоциации 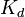 определяется активностями катионов
определяется активностями катионов 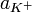 , анионов
, анионов 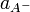 и недиссоциированных молекул
и недиссоциированных молекул 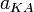 следующим образом:
следующим образом: 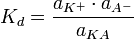
Значение зависит от природы растворённого вещества и растворителя, а также от температуры и может быть определено несколькими экспериментальными методами. Степень диссоциации (α) может быть рассчитана при любой концентрации электролита с помощью соотношения: 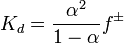 ,где
,где 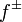 — средний коэффициент активности электролита.
— средний коэффициент активности электролита.
Изотонический коэффициент. Степень электролитической диссоциации, связь с изотоническим коэффициентом. Кажущаяся (Мнимая) степень диссоциации:
Изотонический коэффициент (также фактор Вант-Гоффа; обозначается i) — безразмерный параметр, характеризующий поведение вещества в растворе. Он численно равен отношению значения некоторого коллигативного свойства раствора данного вещества и значения того же коллигативного свойства неэлектролита той жеконцентрации при неизменных прочих параметрах системы: 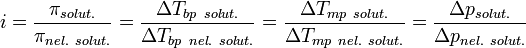 ,
,
где solut. — данный раствор, nel. solut. — раствор неэлектролита той же концентрации, Tbp — температура кипения, а Tmp — температура плавления (замерзания).
Смысл параметра ясен из определения каждого из коллигативных параметров: они зависят от концентрации в растворе частиц растворённого вещества. Неэлектролиты в растворе не диссоциируют, стало быть, каждая молекула неэлектролита образует в растворе лишь одну частицу. В свою очередь, электролиты в растворе под влияниемсольватации частично или полностью распадаются на ионы, образуя при этом несколько частиц на одну диссоциировавшую молекулу. Соответственно, и коллигативные свойства данного раствора (аддитивные величины) зависят от содержания в нём частиц (ионов) каждого типа из тех, которым принадлежат частицы, образовавшиеся в растворе в результате диссоциации исходной молекулы, — раствор представляется как бы смесью растворов каждого из типов частиц. Например, раствор хлорной извести содержит три типа частиц — катионы кальция, хлорид-анионы и гипохлорит-анионы. Итак, изотонический коэффициент показывает, насколько в растворе электролита больше частиц по сравнению с раствором неэлектролита аналогичной концентрации, и связан со способностью вещества распадаться в растворе на ионы, то есть, со степенью диссоциации.
Поскольку сильные электролиты диссоциируют практически полностью, можно было бы ожидать для них изотонический коэффициент, равный количеству ионов (или поляризованных атомов) в формульной единице (молекуле). Однако в действительности этот коэффициент всегда меньше определённого по формуле. Например, изотонический коэффициент для 0,05-моляльного раствора NaCl равен 1,9 вместо 2,0 (для раствора сульфата магния той же концентрации и вовсе i = 1,3). Это объясняеттеория сильных электролитов, разработанная в 1923 году П. Дебаем и Э. Хюккелем: передвижение ионов в растворе затруднено образовавшейся оболочкой сольватации. К тому же, ионы взаимодействуют и между собой: разноимённо заряженные притягиваются, а одноимённо заряженные — отталкиваются; силы взаимного притяжения приводят к образованию групп ионов, перемещающихся по раствору совместно. Такие группы называют ионными ассоциатами или ионными па́рами. Соответственно, раствор ведёт себя так, будто содержит меньше частиц, чем на самом деле, ведь свобода их перемещения ограничена. Наиболее очевиден пример, касающийсяэлектропроводности растворов λ, которая возрастает с разбавлением раствора. Через отношение реальной электропроводности к таковой при бесконечном разбавлении определяют мнимую степень диссоциации сильных электролитов, также обозначаемую через α:
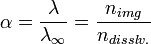 ,
,
где nimg — мнимое, а ndisslv. — реальное количество частиц в растворе.
Физико-химическая теория растворов:
Физико-химическая теория растворов, по существу, заключается уже в учении Бертолле, установившего понятие о химическом равновесии. И современные представления этого рода сводятся к тому, что внутри раствора мы имеем систему подвижного равновесия. Эти представления получили большую определенность благодаря открытию явлений диссоциации. Возникшая во времена Бертолле грань между химическими соединениями и растворами, созданная законом определенных пропорций как характерным признаком химических явлений — устранена. При наступившей диссоциации акт химического взаимодействия непрерывно тянется с изменением действующих масс, подобно тому, как это свойственно силам, ведущим к образованию раствора. Конечный результат действия этих сил — ассоциация, т. е. образование соединений молекул.
Диэлектрическая проницаемость растворителя:
Растворители для растворов электролитов - как правило, полярные жидкости (чистые или смешанные). Чем больше диэлектрическая проницаемость e растворителя, тем значительнее ослабляется сильное электростатическое притяжение противоположно заряженных ионов, что способствует возникновению в растворе ионов. Интенсивное взаимодействие последних с молекулами растворителя приводит к связыванию ионов с молекулами растворителя (см. Сольватация). Важна также способность молекул растворителя выступать в качестве доноров или акцепторов протонов или электронов. В зависимости от этих двух свойств различают четыре группы растворителей: 1) протонные растворители (вода, спирты, карбоновые кислоты и др.), которые являются хорошими донорами протона и обладают высокой диэлектрическая проницаемостью (e > 15); 2) апротонные растворители (некоторые апротонные амиды, кетоны, сульфоксиды и др.), обладающие высокой диэлектрическая проницаемостью, но не обладающие донорно-акцепторными свойствами в отношении протона; 3) электроннодонорные-растворители (например, эфиры); 4) неполярные растворители (сероуглерод, углеводороды), которые обладают низкой диэлектрическая проницаемостью (e < 15) и не обладают донорно-акцепторными свойствами ни по отношению к протону, ни по отношению к электрону.
Классификация электролитов по силе:
Исходя из степени диссоциации все электролиты делятся на две группы
1. Сильные электролиты — электролиты, степень диссоциации которых в растворах равна единице (то есть диссоциируют полностью) и не зависит от концентрации раствора. Сюда относятся подавляющее большинство солей, щелочей, а также некоторые кислоты (сильные кислоты, такие как: HCl, HBr, HI, HNO3).
2. Слабые электролиты — степень диссоциации меньше единицы (то есть диссоциируют не полностью) и уменьшается с ростом концентрации. К ним относят воду, ряд кислот (слабые кислоты), основания p-, d-, и f- элементов.
Между этими двумя группами четкой границы нет, одно и то же вещество может в одном растворителе проявлять свойства сильного электролита, а в другом — слабого.
Активность (Льюис):
Активность компонентов раствора — эффективная (кажущаяся) концентрация компонентов с учётом различных взаимодействий между ними в растворе, то есть с учётом отклонения поведения системы от модели идеального раствора.
Активность была предложена в 1907 году Льюисом как новая переменная, применение которой вместо концентрации позволяет использовать для описания свойств реальных растворов относительно простые уравнения, полученные для идеальных систем. Альтернативой этому пути является использование более сложных уравнений, учитывающих взаимодействие между частицами (см., например, уравнение Ван-дер-Ваальса).
Активность отличается от общей концентрации на некоторую величину. Отношение активности (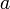 ) к общей концентрации вещества в растворе называется коэффициентом активности:
) к общей концентрации вещества в растворе называется коэффициентом активности:
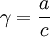
Коэффициент активности служит мерой отклонения поведения раствора (или компонента раствора) от идеального. Отклонения от идеальности могут быть обусловлены различными химическими и физическими причинами — дипольные взаимодействия, поляризация, образование водородных связей, ассоциация, диссоциация, сольватация и др.
Понятие о теории Дебая-Хюккеля:
Теориясильных электролитов Дебая—Хюккеля — статистическая теория разбавленных растворов сильных электролитов, согласно которой каждый ион действием своего электрического заряда поляризует окружение и образует вокруг себя некоторое преобладание ионов противоположного знака — так называемую ионную атмосферу.
В отсутствие внешнего электрического поля ионная атмосфера имеет сферическую симметрию, и её заряд равен по величине и противоположен по знаку заряду создающего её центрального иона. В этой теории не уделено почти никакого внимания образованию пар противоположно заряженных ионов путём непосредственного взаимодействия между ними.
Ионная сила:
Ионная сила раствора — мера интенсивности электрического поля, создаваемого ионами в растворе. Полусумма произведений из концентрации всех ионов в растворе на квадрат их заряда. Формула впервые была выведена Льюисом:
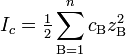 ,
,
где c B — молярные концентрации отдельных ионов (моль/л), z B заряды ионов
Суммирование проводится по всем типам ионов, присутствующих в растворе. Если в растворе присутствуют два или несколько электролитов, то вычисляется общая суммарная ионная сила раствора.
Это верно для раствора любого сильного электролита, состоящего из однозарядных ионов. Для электролитов, в которых присутствуют многозарядные ионы, ионная сила обычно превышает молярность раствора.
Ионная сила раствора имеет большое значение в теории сильных электролитов Дебая — Хюккеля. Основное уравнение этой теории (предельный закон Дебая — Хюккеля) показывает связь между коэффициентом активности иона zi и ионной силы раствора I в виде:
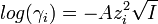 ,
,
где γ — коэффициент активности, А — постоянная, не зависящая от заряда иона и ионной силы раствора, но зависящая от диэлектрической постоянной растворителя и температуры.
Солевой эффект:
В общем смысле солевой эффект - это эффект изменения как течения реакции (например, выход продуктов, стерехимию, регеоселективность), так и положения химического равновесия, наблюдаемый при добавлении в реакционную среду химически инертных солей.
Солевой эффект - это явление увеличения ПР осадка при добавлении сильного электролита (не содержащего одноимённых ионов).
Кислоты и основания. Кислотно-основные реакции. Протолитическая теория Бренстеда-Лаури. Протолитические превращения в растворах электролитов. Сопряженные протолитические пары кислота/основание. Теория Лукса–Флуда. Теория Усановича. Электронная теория кислотно-основных реакций Льюиса. Теория ЖМКО (Пирсон).
Кислоты и основания:
Кислоты — сложные вещества, в состав которых обычно входят атомы водорода, способные замещаться на атомы металлов, и кислотный остаток. Водные растворы кислот имеют кислый вкус, обладают раздражающим действием, способны менять окраску индикаторов, отличаются рядом общих химических свойств.
Основания — класс химических соединений.
· Основания (основные гидроксиды) — сложные вещества, которые состоят из атомов металла или иона аммония и гидроксогруппы (-OH). В водном растворедиссоциируют с образованием катионов и анионов ОН−. Название основания обычно состоит из двух слов: «гидроксид металла/аммония». Хорошо растворимые в воде основания называются щелочами.
· Согласно протонной теории кислот и оснований, основания — один из основных классов химических соединений, вещества, молекулы которых являются акцепторами протонов.
· В органической химии по традиции основаниями называют также вещества, способные давать аддукты («соли») с сильными кислотами, например, многиеалкалоиды описывают как в форме «алкалоид-основание», так и в виде «солей алкалоидов».
Кислотно-основные реакции. Протолитическая теория Бренстеда-Лаури. Протолитические превращения в растворах электролитов. Сопряженные протолитические пары кислота/основание:
Протолитическая (протонная) теория кислот и оснований была предложена в 1923 году независимо друг от друга датским учёным Й. Бренстедом и английским учёным Т. Лаури. В ней понятие о кислотах и основаниях было объединено в единое целое, проявляющееся в кислотно-основном взаимодействии: А  В + Н+ (А - кислота, В - основание). Согласно этой теории кислотами являются молекулы или ионы, способные быть в данной реакции донорами протонов, а основаниями являются молекулы или ионы, присоединяющие протоны (акцепторы). Кислоты и основания получили общее название протолитов.
В + Н+ (А - кислота, В - основание). Согласно этой теории кислотами являются молекулы или ионы, способные быть в данной реакции донорами протонов, а основаниями являются молекулы или ионы, присоединяющие протоны (акцепторы). Кислоты и основания получили общее название протолитов.
Сущностью кислотно-основного взаимодействия является передача протона от кислоты к основанию. При этом кислота, передав протон основанию, сама становится основанием, так как может снова присоединять протон, а основание, образуя протонированную частицу, становится кислотой. Таким образом, в любом кислотно-основном взаимодействии участвуют две пары кислот и оснований, названные Бренстедом сопряженными: А1 + В2 А2 + В1.
Одно и то же вещество в зависимости от условий взаимодействия может быть как кислотой, так и основанием (амфотерность). Например, вода при взаимодействии с сильными кислотами является основанием: H2O + H+ H3О+, а реагируя с аммиаком, становится кислотой: NH3 + H2O NH4+ + OH−.
Теория Лукса–Флуда:
Для описания кислотно-основных взаимодействий в расплавах используется теория Лукса-Флуда, в соответствии с которой кислота – акцептор оксид-ионов (SiO2), а основание – донор оксид-ионов (CaO):CaO + SiO2 = CaSiO3.
Теория Усановича:
Наиболее общая теория кислот и оснований была сформулирована М. Усановичем в 1939 году. В основе теории лежит представление о том, что всякое кислотно-основное взаимодействие — это реакция солеобразования. Согласно этой теории «кислота — это частица, которая может отщеплять катионы, включая протон, или присоединять анионы, включая электрон. Основание — частица, которая может присоединять протон и другие катионы или отдавать электрон и другие анионы» (формулировка 1964 г.). В отличие от Льюиса Усанович в основе понятий «кислота» и «основание» использует знак заряда частицы, а не строение электронной оболочки.
По Усановичу, в реакции гидролиза SO3 + 2H2O H3O+ + HSO4- — вода, отдавая анион OH-, является основанием, а триоксид серы, присоединяя этот анион — кислотой, аналогично в реакции: SnCl4 + 2KCl K2SnCl6 — тетрахлорид олова, присоединяющий анионы хлора, выступает в роли кислоты. Таким образом, данная формулировка кислот и оснований позволяет отнести к кислотно-основным взаимодействиям и все окислительно-восстановительные реакции.
Теория Усановича фактически отменяет один из основополагающих принципов классической химии — представления о классах кислот и оснований: «кислоты и основания — это не классы соединений; кислотность и основность — это функции вещества. Будет ли вещество кислотой или основанием, зависит от партнера».
К недостаткам теории Усановича относят её слишком общий характер и недостаточно чёткую определённость формулировки понятий «кислота» и «основание». К недостаткам относят также то обстоятельство, что она не описывает неионогенные кислотно-основные превращения. Наконец, она не позволяет делать количественные предсказания.
Электронная теория кислотно-основных реакций Льюиса:
В теории Льюиса (1923 г.) на основе электронных представлений было ещё более расширено понятие кислоты и основания.Кислота Льюиса — молекула или ион, имеющие вакантные электронные орбитали, вследствие чего они способны принимать электронные пары. Это, например, ионы водорода – протоны, ионы металлов (Ag+, Fe3+), оксиды некоторых неметаллов (например, SO3, SiO2), ряд солей (AlCl3), а также такие вещества как BF3, Al2O3. Кислоты Льюиса, не содержащие ионов водорода, называются апротонными. Протонные кислоты рассматриваются как частный случай класса кислот. Основание Льюиса — это молекула или ион, способные быть донором электронных пар: все анионы, аммиак и амины, вода, спирты, галогены. Примеры химических реакций между кислотами и основаниями Льюиса:
· AlCl3 + Cl− → AlCl4−
· BF3 + F− → BF4−
· PCl5 + Cl− → PCl6−.
Теория ЖМКО (Пирсон):
Под этой аббревиатурой скрывается теория жестких и мягких кислот и оснований, сформулированная в 60-х годах Р. Пирсоном и являющаяся своеобразным развитием теории Усановича. Но прежде чем изложить основные положения этой теории, следует познакомиться с таким важным свойством нейтральных и заряженных частиц, как поляризуемость. Каждый атом, молекула или ион окружены электронным облаком — пространством, в котором движутся по орбиталям электроны. Под влиянием внешнего поля форма этого облака может изменяться. Это свойство называют поляризуемостью. Так вот, у различных частиц способность к поляризуемости неодинакова. Атомы (например, аргона или неона) или ионы (например, фтора или калия), обладающие завершенной электронной оболочкой, которая к тому же недалеко расположена от ядра и поэтому достаточно сильно притягивается к нему, обладают низкой поляризуемостью. Атомы же или ионы с незавершенной электронной оболочкой, либо частицы, у которых внешняя орбиталь далека от ядра, достаточно легко изменяют форму электронного облака и поэтому обладают высокой поляризуемостью.
Теория ЖМКО делит, как это уже следует из названия, все кислоты и основания на жесткие и мягкие. Согласно определениям теории ЖМКО, к жестким основаниям относят донорные частицы (т. е. частицы, которые могут делиться с другими частицами своими электронами либо даже полностью отдавать их), обладающие высокой электроотрицательностью, низкой поляризуемостью и трудно окисляющиеся. Многие анионы могут служить примером жестких оснований: ОН-, F-, S042-; жесткими основаниями могут быть и нейтральные молекулы, например NH3, CH3NH2. Донорные частицы с низкой электроотрицательностью и высокой поляризуемостью составляют класс мягких оснований. Как правило, эти частицы сравнительно легко окисляются. В качестве примера здесь могут быть названы I-, SCN-, (СН3)3Р, С6Н6.
Жесткие кислоты, по Пирсону, это акцепторные (т. е. обладающие склонностью к электрону) частицы с низкой поляризуемостью. К ним относят, например, Н+, Li+, Na+, BF3. Наконец, тип мягких кислот составляют акцепторные частицы с высокой поляризуемостью. Это Ag+, GaCl3 и др.
Суть теории ЖМКО состоит в том, что жесткие кислоты преимущественно реагируют с жесткими же основаниями, а мягкие кислоты — с мягкими основаниями. Этот стержневой принцип не свободен от ряда исключений, но тем не менее является достаточно общим, в чем читатель может убедиться, составляя соответствующие сочетания.
Последовательно применяя принцип теории ЖМКО ко всем возможным частицам, можно прийти к заключению, что наиболее жесткая кислота — это позитрон, а наиболее жесткое основание — электрон.
Химические равновесия в водных растворах слабых электролитов. Закон разбавлений Оствальда. Диссоциация (ионизация) воды (автопротолиз). Ионное произведение воды. Водородный и гидроксидный показатели.
Химические равновесия в водных растворах слабых электролитов:
Химическим равновесием называют такое состояние системы реагирующих веществ, при котором скорости прямой и обратной реакций равны.
Для количественной характеристики равновесия используют константы равновесия, выражаемые через равновесные концентрации компонентов реакции. Так, для системы m A+ n B↔ p C+ q D константу равновесия (K) вычисляют в соответствии с законом действующих масс (З.Д.М.) по формуле
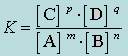
Численное значение константы равновесия дает возможность установить:
a. направление протекания химической реакции;
b. полноту протекания, т. е. является ли реакция обратимой или практически необратимой (чем больше численное значение K, тем полнее протекает реакция).
Формула в таком виде справедлива только для слабых электролитов и очень разбавленных растворов сильных электролитов.
Закон разбавлений Оствальда:
Закон разбавления Оствальда — соотношение, выражающее зависимость эквивалентной электропроводности разбавленного раствора бинарного слабого электролита от концентрации раствора:
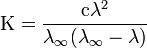
Здесь К — константа диссоциации электролита, с — концентрация, λ и λ∞ — значения эквивалентной электропроводности соответственно при концентрации с и при бесконечном разбавлении. Соотношение является следствием закона действующих масс и равенства: 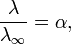
где α — степень диссоциации.
Диссоциация (ионизация) воды (автопротолиз):
Чистая вода, хоть и плохо (по сравнению с растворами электролитов), но может проводить электрический ток. Это вызвано способностью молекулы воды распадаться (диссоциировать) на два иона которые и являются проводниками электрического тока в чистой воде (ниже под диссоциацией подразумевается электролитическая диссоциация - распад на ионы): H2O ↔ H+ + OH-
Вследствие очень малого количества продиссоциированных молекул концентрацию [H2O] можно принять равной общей концентрации воды, а общую концентрацию воды в разбавленных растворах как величину постоянную: [H2O]=1000(г/л)/18(г/моль)=55,6 моль/л.
Так как, при определенной температуре, величины используемые в расчете ионного произведения воды (K, [H2O]) постоянны, значение ионного произведения воды так же постоянно. А поскольку при диссоциации молекулы воды образуется одинаковое количество ионов [H+] и [HO-], получается что для чистой воды концентрации [H+] и [HO-] будут равны 10-7 моль/л. Из постоянства ионного произведения воды следует, что если количество ионов H+ становится больше, то количество ионов HO- становится меньше. Например, если к чистой воде добавить сильную кислоту HCl, она как сильный электролит вся продиссоциирует на H+ и Cl-, в результате концентрация ионов H+ резко увеличится, и это приведет к увеличению скорости процесса противоположного диссоциации, так как она зависит от концентраций ионов H+ и OH-.
Ионное произведение воды́ — произведение концентраций ионов водорода Н+ и ионов гидроксила OH− в воде или в водных растворах, константа автопротолиза воды.
Водородный и гидроксидный показатели:
Водородный показатель, pH — мера активности (в очень разбавленных растворах она эквивалентна концентрации) ионов водорода в растворе, и количественно выражающая его кислотность, вычисляется как отрицательный (взятый с обратным знаком) десятичный логарифм активности водородных ионов, выраженной в молях на литр: 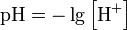
Гидроксидный показатель – это отрицательный десятичный логарифм концентрации иона ОН-:
рОН=-lg[ОН-],
рН + рОН=14.
При рН >7 среда щелочная, рН=7 – нейтральная, рН<7 – кислая.