 2018-02-13
2018-02-13 2574
2574Галогенпроизводные углеводородов.
Очень большое значение в органической химии имеют галогенпроизводные углеводородов, которые вследствие своей высокой реакционной способности используют как исходные реагенты для синтеза многих других классов органических соединений. По сути дела галогенпроизводные являются промежуточным звеном в генетической связи между классами органических соединений, где первым звеном стоят сами углеводороды, вторым чаще всего их галогенпроизводные, которые превращаются в другие классы.
За неимением времени не будем рассматривать ни методы получения, ни свойства галогенпроизводных. Методы получения уже неоднократно встречались, а из множества свойств выбираем два наиболее важных типа реакций – нуклеофильное замещение и отщепление и рассматриваем их механизмы.
Нуклеофильное замещение.
Вы уже имеете представление об SN1 и SN2 реакциях (см. раздел 6., здесь мы остановимся на двух моментах – влияние структуры реагентов на механизм и скорость, а также стереоспецифичность этих реакций.
12.3.1.Влияние структуры реагентов.
По своей реакционной способности в нуклеофильных реакциях галогенпроизводные располагаются в ряд:
R-F << R-Cl < R-Br < R-I.
Причина – уменьшение прочности связи С-Hal (и увеличение ее поляризуемости).
Скорость нуклеофильного замещения зависит от структуры радикала R, что можно продемонстрировать при помощи графика:
 |
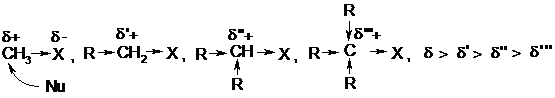 |
В реакциях СН3Х и RCH2X, как отмечалось выше, карбокатионы не могут образовываться, ибо они очень нестабильны, т.е. возможен только SN2 механизм. При этом скорость реакции уменьшаются при переходе от СН3 к первичным и вторичным радикалам вследствие их + І-эффектов, уменьшающих d+ заряд на реакционном центре:
Тем самым понижается скорость реакции с нуклеофилом Nud-. Кроме того, атаке препятствует и стерическое отталкивание радикалов R, поэтому SN2 механизм для третичных производных невозможен, здесь протекают только SN1 реакции по двум причинам:
1) образуется стабилизированный за счет гиперконьюгации карбокатион R3C+,
2) отсутствуют стерические препятствия атаке Nu на этот карбокатион, имеющий плоскую структуру вследствие sp2-гибридного состояния атома углерода.
12.3.2. Стереохимия реакций замещения.
12.3.2.1. Стереохимия реакций SN1-замещения.
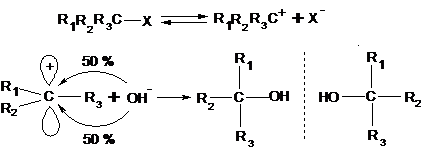 |
Скорость реакции определяется самой медленной первой стадией, поэтому W = k [RX]. Cтереохимия реакции:
рацемат
Образуется оптически неактивная эквимолекулярная рацемическая смесь энантиомеров (оптических антиподов).
Таким образом, при SN1-замещении всегда образуется оптически неактивный рацемический продукт реакции.
12.3.2.2. Стереохимия реакций SN2-замещения.
Процесс одностадийный, а скорость реакции определяется уравнением W = k [RX][Nu]. Пример:

Здесь имеет место тыловая атака (Вальденовское обращение конфигурации):

S-конфигурация R-конфигурация
12.3.2.3. Стереохимия SN1-замещения с участием ионных пар.
 |
В ряде малополярных растворителей образование свободных ионов затруднено, поэтому диссоциация RX останавливается на образовании ионных пар – контактных (КИП) и сольватно-разделенных (СРИП):
 |
Анион в ионной паре частично мешает атаке нуклеофила на катион с той стороны, с которой он находится:
Поэтому идет частичная рацемизация наряду с частичным Вальденовским обращением.
 |
В ряде случаев растворитель, например Н2О, проникает между ионами и образует СРИП:
Если же эта молекула воды (или другого растворителя, например, спирта) выступает в качестве нуклеофильного реагента, то имеет место сохранение конфигурации.
12.4. Побочные реакции при SN1-замещении.

В этом случае протекает три параллельных реакции:
 |
В промежуточном катионе два реакционных центра – на атоме углерода и на атоме водорода в b-положении:
Нуклеофил может атаковать оба центра: при атаке на углерод образуется продукт замещения, при атаке на водород – продукт отщепления. Реакции замещения преобладают в водной среде, а отщепления преобладают в менее полярной спиртовой среде.
Реакции отщепления.
Пример:
спирт
 (СН3)3СCl + KOH KCl + H2O + (CH3)2C=CH2.
(СН3)3СCl + KOH KCl + H2O + (CH3)2C=CH2.
Имеется три типа механизмов реакции отщепления.
12.5.1. Е1 механизм.

Пример реакции, катализируемой этилатом натрия в этиловом спирте и протекающей по этому механизму:
Первая стадия самая медленная, поэтому скорость такой реакции определяется уравнением:
W = k [RX].
Преимущественно по этому механизму протекают реакции отщепления с участием третичных алкилгалогенидов, предварительно распадающихся, как и при нуклеофильном SN1 замещении, на ионы, причем, образующийся карбокатион является общим нестабильным промежуточным продуктом для обоих процессов. Поэтому, как уже отмечалось в разделе 12.4, для третичных алкилгалогенидов реакции отщепления и замещения протекают параллельно.
12.5.2. Е2 механизм.
Примеры реакций:
вода
 СН3-СН2-N+(CH3)3×Cl- + KOH KCl + H2O + CH2=CH2 + N(CH3)3;
СН3-СН2-N+(CH3)3×Cl- + KOH KCl + H2O + CH2=CH2 + N(CH3)3;
спирт
 СН3-СН2-СНBr-СН3 + KOR KBr + HOR + СН3-СН=СН-СН3
СН3-СН2-СНBr-СН3 + KOR KBr + HOR + СН3-СН=СН-СН3
 |
Механизм одностадийный, поскольку уходящая группа (триметиламин в первой реакции и Br- во второй) отщепляется одновременно с протоном в переходном состоянии:
Поэтому скорость реакции описывается бимолекулярным уравнением:
W = k[RO-][RX].
12.5.3. Мономолекулярное отщепление сопряженным основанием (Е1сВ).
Этот механизм осуществляется, если уходящая группа очень трудно отщепляется. Пример такой реакции в метаноле:
 СН3ОNa + H-CCl2-CF2-F СН3ОН + Cl2С=CF2 + NaF
СН3ОNa + H-CCl2-CF2-F СН3ОН + Cl2С=CF2 + NaF
 |
Механизм:
В качестве промежуточного продукта здесь образуется карбоанион. Этот механизм весьма редко встречается, а подавляющая масса реакций отщепления в зависимости от строения субстрата идут либо по Е2, либо по Е1 механизму.
12.5.4.Соотношение процессов отщепления и замещения в водной и
спиртовой среде.
Как уже отмечалось в разделе 12.4, алкилгалогениды в водных растворах щелочей преимущественно вступают в реакции замещения, тогда как в спиртовых растворах в присутствии таких сильных оснований, как алкоголяты металлов (см. лекцию № 13), преимущественно идут реакции отщепления
Объяснить направление реакции в разных средах можно, исходя из концепции жестких и мягких кислот и оснований, в соответствии с которой мягкие кислоты и основания, обладающие высокой поляризуемостью, реагируют между собой, а жесткие кислоты и основания, уже поляризованные и поэтому имеющие низкую прляризуемость, реагируют между собой. Примеры взаимодействий:
| Тип взаи- Модейству-ющих час- Тиц | Кислотный центр в Н-С-С+ Н+ С=С | Основание | Растворитель | Механизм |
| Жесткие | Н+ | RO- | Спирт | Е |
| Мягкие | С+ | HO- | Вода | SN |
В спирте протекание реакций отщепления обеспечивают алкоголят анионы, которые являются более жесткими (более сильные вследствие положительного индуктивного эффекта углеводородных радикалов) основаниями, чем гидроксид анион (более мягок, поскольку в воде легче поляризуется), и реагируют преимущественно с жесткой кислотой Н+ (подобное реагирует с подобным). В воде же мягкие основания HO- реагируют с мягким кислотным центром С+, вследствие чего идет реакция замещения.

В спиртовом растворе КОН в отличие от водных растворов того же КОН также преимущественно идет отщепление, а не замещение. Такой факт можно объяснить тем, что более основные, чем вода (опять же вследствие упомянутых выше индуктивных эффектов углеводородных радикалов), молекулы спирта, сольватируя гидроксид анион, усиливают основность последнего настолько, что он становится достаточно жестким, чтобы реагировать не с положительно заряженным углеродом, а с наиболее кислым водородом в b-положении к отщепляемой группе Х:
12.5.5. Правило отщепления Зайцева.
 |
При отщеплении галогеноводородов, когда отщепление галогенид аниона (уходящего нуклеофила) опережает отщепление протона, то протон преимущественно отщепляется от наименее гидрогенизированного атома углерода. Это объясняется тем, что при отщеплении по Е1 и Е2 механизмам с переходным состоянием типа (12.І) должен образовываться наиболее стабильный, т.е. наиболее замещенный, алкен (стабилизация за счет гиперконьюгации). Например, при отщеплении HBr от 3-бром-2,3-диметилпентана по Е1-механизму вследствие отщепления протона от трех соседних групп (СН, СН2 или СН3) в промежуточном карбокатионе [(СН3)2СН]C+[CH3][CH2CH3] образуются соединения, выход которых уменьшается в ряду (ибо в этом же ряду уменьшается их стабильность):
Стабильность алкенов определяется числом алкильных заместителей при двойной связи, что в свою очередь обусловлено гиперконьюгацией. Например, в случае пропилена гиперконьюгацию можно описать следующими резонансными формулами:
 |
И чем больше резонансных структур описывает строение вещества, тем оно стабильнее. Поэтому замещенные этилены можно расположить в ряд в порядке увеличения их стабильности: СН3-СН=СН2 (4 структуры) < (СН3)2С=СН2 или СН3-СН=СН-СН3 (7 структур) < (СН3)2С=СН-СН3 (10 структур) <
< (СН3)2С=С(СН3)2 (13 структур).
 |
При отщеплении НCl от 2-хлорбутана выходы уменьшаются в ряду:
транс-бутен-2 цис-бутен-2 бутен-1
Выходы геометрических изомеров объясняются стабильностью конформаций исходного реагента:
 |
заторможенная скошенная
анти-конформация син-конформация
Легче образуется транс-изомер, ибо конформация А, из которой он образуется (кружками обозначены атомы, которые отщепляются), стабильнее, чем конформация В, в которой метильные группы отталкиваются. Кроме того, в конформации А выполняются требования, чтобы отщепляемые атомы лежали в одной плоскости, ибо р-орбитали атомов углерода, от которых атомы водорода и хлора отщепляются, должны лежать в одной плоскости, чтобы, перекрываясь, образовывать p-связь.
Лекция № 13.
ГИДРОКСИЛСОДЕРЖАЩИЕСОЕДИНЕНИЯ: СПИРТЫ И ФЕНОЛЫ.
13.1. Введение.
 |
Гидроксилсодержащие соединения делятся на спирты, в которых гидроксильные группы связаны с алкильным радикалом, и фенолы, где гидрокси-группа непосредственно связана с бензольным (ароматическим) ядром:
Метанол фенол, гидроксибензен
По количеству гидроксильных групп спирты и фенолы делятся на одноатомные (примеры даны выше), двухатомны, трехатомные, т.е. полиатомные. Примеры:

Этиленгликоль глицерин пирокатехин пирогаллол
Этандиол-1,2 пропантриол-1,2,3 1,2-дигидроксибензол 1,2,3-тригидрокси-
бензолдиол-1,2 бензен
В зависимости от природы углеводородного скелета спирты делятся на:
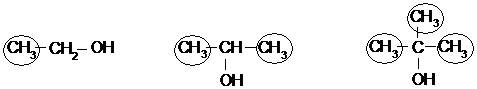
первичный вторичный третичный.
13.2. Одноатомные насыщенные спирты и фенолы.
13.2.1. Изомерия и номенклатура.
 |
Изомерия спиртов и фенолов, как и других классов соединений, обусловлена изомерией скелета и положением функциональной группы. Общие правила номенклатуры вам уже известны. Приведу примеры с применением трех номенклатур:
ИЮПАК пропанол-1; пропанол-2; 2-метилпропанол-2; 2-метилпропанол-1
Радикально- пропиловый изопропило- трет-бутиловый изобутиловый
функцио- спирт вый спирт спирт спирт
нальная
Карбинольная этилкарби- диметил- тримпетилкар- изопрпилкарбинол
нол карбинол бинол
По карбинольной номенклатуре все спирты считают производными карбинола, т.е. метилового спирта СН3-ОН.
13.2.2. Методы получения.
13.2.2.1. Гидратация алкенов (уже рассмотрено).
13.2.2.2. Гидролиз алкил- и арилгалогенидов.
Гидролиз алкилгалогенидов уже рассматривался, гидролиз арилгалогенидов идет с трудом вследствие сопряжения галогена с кольцом, приводящего к упрочнению связи С-Гал:
250oC/Cu
 С6Н5Сl + NaOH С6Н5OH + NaCl
С6Н5Сl + NaOH С6Н5OH + NaCl
 |
13.2.2.3. Магнийорганический синтез (в диэтиловом эфире)
Таким образом, при взаимодействии магнийорганических соединений с формальдегидом образуется первичный спирт А, с любым альдегидом – вторичный спирт В, с кетоном (в данном случае ацетоном) – третичный спирт С.

13.2.2.4. Получение фенола из кумола.
13.2.3. Физические свойства. Ассоциация.
Для спиртов и фенолов характерна ассоциация за счет водородных связей, которые повышают температуры кипения и плавления. Поэтому метанол (М=32) – жидкость, тогда как этан (М=30) – газ. Фенолы по той же причине – твердые вещества.
13.2.4. Химические свойства.
Химические свойства определяются наличием полярных С-О и О-Н связей, причем, О-Н полярнее, чем С-О, поскольку различие электроотрицательностей в первом случае больше, чем во втором.
Для спиртов характерны две группы реакций: с разрывом связей О-Н и с
разрывом связей С-О.
13.2.4.1. Реакции с расщеплением О-Н связей.
13.2.4.1.1. Кислотно-основные свойства.
Кислотные свойства спиртов слабее, чем воды, вследствие + І-эффекта радикала, увеличивающего отрицательный заряд на кислороде и тем самым упрочняющего связь О-Н (сильнее притягивает протон). Ряд кислотности:
 R Od--Hd+ < H-OH < C6H5-O-H < H2CO3.
R Od--Hd+ < H-OH < C6H5-O-H < H2CO3.
 |
Более сильные кислотные свойства фенолов, чем Н2О, объясняются взаимным влиянием кольца и гидроксила:
За счет сопряжения здесь, с одной стороны, усиливаются кислотные свойства, но, с другой, увеличивается реакционная способность кольца к электрофильному реагенту Е+ (взаимное влияние бензольного ядра и гидроксильной группы). Поскольку спирт по кислотности слабее воды, он не может вытеснять ее из щелочи, благодаря чему спирт со щелочью не реагирует. В случае фенола дело обстоит наоборот – он реагирует со щелочью, вытесняя из нее более слабую воду:
 С6Н5ОН + NaOH С6Н5ОNa + H2O.
С6Н5ОН + NaOH С6Н5ОNa + H2O.
Кислотность спиртов все же проявляется в их взаимодействии со щелочными металлами, в результате чего образуются алкоголяты этих металлов:
 2R-OH + 2Na 2R-ONa + H2.
2R-OH + 2Na 2R-ONa + H2.
Вода, как более сильная кислота, вытесняет более слабый спирт из его алкоголята:
 RONa + H2O ROH + NaOH.
RONa + H2O ROH + NaOH.
13.2.4.1.2. Реакции этерификации.
Это реакции замещения кислого атома водорода спирта на ацильный остаток карбоновых кислот:

 R-*O-H + HO-CO-R¢ H2O + R-*O - CO-R¢.
R-*O-H + HO-CO-R¢ H2O + R-*O - CO-R¢.
Реакция обратима. Если кислород спирта пометить изотопом 18О (в уравнении *O), то метка попадает в эфир, ибо спирт выступает как нуклеофил с реакционнім центром на кислороде, а кислота как электрофильный реагент. Реакции катализируются сильными кислотами (H2SO4, H3PO4).
13.2.4.1.3. Реакции окисления и дегидрирования.
 |
Реакции дегидрирования могут идти как в отсутствии кислорода (катализ Cu), так и в присутствии кислорода:
Из вторичных спиртов образуются кетоны:
300оС/Cu
 R2CH-OH R2C=O + H2.
R2CH-OH R2C=O + H2.
Реакции дегидрирования в присутствии кислорода называются реакциями окислительного дегидрирования (или просто окисления). Спирты могут окисляться KMnO4, K2Cr2O7 и кислородом воздуха в присутствии Cu, первичные спирты окисляются до альдегидов, вторичные – до кетонов. Третичные спирты окисляются значительно труднее с расщеплением углеродной цепочки.
 |
Фенолы окисляются в присутствии хромовой смеси (H2SO4 + K2Cr2O7):
гидрохинон хинон
13.2.4.2. Реакции с разрывом связей С-О.
13.2.4.2.1. Реакции замещения гидроксила на галоген:
R-OH + HCl R-Cl + H2O.
Гидроксильная группа – плохая уходящая группа, намного более слабый по основности галогенид-анион не может ее вытеснить, поэтому здесь необходим катализ кислотами:
 R-OH + HCl R-+OH2 + Cl-
R-OH + HCl R-+OH2 + Cl-
 R-+OH2 R+ + OH2
R-+OH2 R+ + OH2
 R+ + Cl- RCl
R+ + Cl- RCl

Здесь SN1 механизм, ибо наблюдается перегруппировка, например, в случае неопентилового спирта:
13.2.4.2.2. Образование простых эфиров (этеров).
При нагревании избытка спирта с серной кислотой при достаточно низкой температуре идет реакция:
 C2Н5-ОН + НО-С2Н5 Н2О + C2Н5-О-С2Н5.
C2Н5-ОН + НО-С2Н5 Н2О + C2Н5-О-С2Н5.
Механизм:
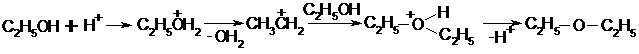
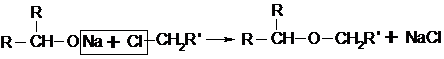
Легче всего реакция идет с первичными спиртами, труднее с вторичными и третичными, поскольку последние вступают в реакции элиминирования. В этом случае реакцию проводят с участием алкоголятов спиртов (сильные нуклеофилы) с алкилгалогенидами:
 |
Но и здесь имеют место трудности, связанные с конкуренцией процессов замещения и отщепления. Здесь в качестве реагентов нельзя брать третичные алкилгалогениды типа R3CX, поскольку они легче вступают в реакции отщепления, чем замещения. С другой стороны, поскольку для третичных алкоголятов R3CО-, обладающих большим объемом и высокой основностью, при атаке на субстрат доступ к a-углероду более затруднен, чем к периферийному водороду в b-положении, то также преобладает отщепление:
13.2.4.3. Реакции дегидратации.
 |
Выше рассматривались межмолекулярные реакции отщепления воды от спиртов с образованием простых эфиров. Внутримолекулярная дегидратация спиртов приводит к алкенам (условия такой дегидратации см. в разделе 7.2.2 лекции № 7):
 |
Легче всего идут реакции с участием третичных спиртов (механизм Е1):
Отщепление воды от спиртов определяется правилом Зайцева: при отщеплении воды водород отщепляется преимущественно от наименее гидрогенизированного соседнего атома углерода:


При имеющем здесь место Е2-механизме отщепления образование наиболее стабильного алкена осуществляется следующим образом:
В случае первичных спиртов реакции отщепления сопровождаются процессами внутримолекулярных перегруппировок (по сути внутримолекулярное нуклеофильное замещение с участием гидрид-анионов в данном случае), которые приводят к образованию трех алкенов, выход которых в соответствии с их стабильностью уменьшается в ряду В > С > А:

13.2.4.4. Специфические свойства фенолов.
13.2.4.4.1. Образование фенолята железа.
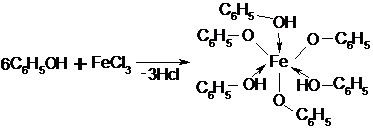 |
С треххлористым железом фенол дает фиолетовое окрашивание. Это характерные реакции на фенолы и енолы:
13.2.4.4.2. Реакции электрофильного замещения.
Фенольный гидроксил сильно активирует бензольное ядро к реакциям электрофильного замещения в о- и п-положения, что следует из резонансных структур, представленных в разделе 13.2.4.1.1. Поэтому фенол с бромной водой в отсутствие катализа сразу же образует 2,4,6-трибромфенол в виде белого осадка, который при избытке брома окисляется до кетона:
 |
Фенолы очень легко нитруются. В разбавленной азотной кислоте образуются моно нитропроизводные (о- и п-нитрофенолы), из смеси которых с водяным паром отгоняется о-нитрофенол (объясните, почему). При нитровании смесью концентрированных азотной и серной кислот сразу вводится три нитрогруппы с образованием 2,4,6-тринитрофенола или пикриновой кислоты, которая является бризантным взрывчатым веществом:
 |
В концентрированной азотной кислоте помимо нитрования происходят процессы окисления фенола и осмоления, поэтому пикриновую кислоту в промышленности получают по схеме:
Получающаяся при сульфировании фенола 2,4-фенолдисульфоновая кислота устойчива к окислительному действию концентрированной азотной кислоты, в которой ее и нитруют. При этом на нитро-группы замещается не только водород в 6-положении, но и обе сульфо-группы.
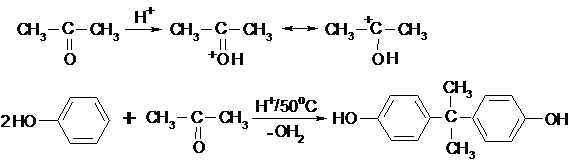 |
13.2.4.4.3. Реакции С-алкилирования (катализатор H2SO4).
Пример – реакция с ацетоном. Электрофил – протонированный ацетон:
2,2-бис-(п-гидроксифенил)пропан
Полученное по этой реакции соединение используется для синтеза красителей, ПАВ, антиоксидантов, полимерных материалов и т.д.
 |
Реакции С-алкилирования происходят при конденсации фенола с формальдегидом. Реакции идут как в кислой, так и в щелочной среде: в шелочной активируется фенол, превращающийся в более активный фенолят, в кислой - альдегид:
В щелочной среде замещение (конденсация) идет как в о- так и в п-положение, поэтому образуются сшитые цепочки. В зависимости от условий (среда и температура) можно получать различные виды смол.

13.2.4.4.4. Реакция Кольбе (СО2 под давлением)
Здесь мягкая кислота (углерод в углекислом газе) атакует мягкое основание (углерод в о-положении фенолята натрия).
 |
13.2.4.4.5. Каталитическое гидрирование.
Этим способом синтезируют циклогексанол – сырье для получения адипиновой кислоты.
Лекция № 14.
СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ несколько ГИДРОКСИЛьных групп. ПРОСТЫЕ ЭФИРЫ.
Гликоли.
14.1.1. Номенклатура.
 |
Это двухатомные спирты. По радикально-функциональной номенклатуре к названию алкена соответствующей длины добавляется окончание «гликоль», они делятся на a-, b-, g- и т- и т.д. гликоли:
a-пропиленгликоль b-пропиленгликоль
пропандиол-1,2 пропандиол-1,3
14.1.2. Получение.
13.1.2.1. Гидролиз дигалогенпроизводных.
 Cl-CH2-CH2-Cl + 2 H2O 2 HCl + HO-CH2-CH2-OH (этиленгликоль).
Cl-CH2-CH2-Cl + 2 H2O 2 HCl + HO-CH2-CH2-OH (этиленгликоль).
14.1.2.2. Окисление алкенов по реакции Вагнера (уже давал).
14.1.3. Химические свойства.
13.1.3.1. Кислотные свойства.
 |
Соседняя НО- группа проявляет -І эффект, вследствие чего a-гликоли кислее воды, реагируют со щелочами, образуя гликоляты. С гидроксидом меди гликоли образуют комплексные соединения – клешневидные хелаты (раствор синего цвета):
14.1.3.2. Дегидратация.
 |
14.1.3.2.1. Внутримолекулярная дегидратация.
 |
14.1.3.2.. Межмолекулярная дегидратация.
14.1.3.3. Реакции окисления.
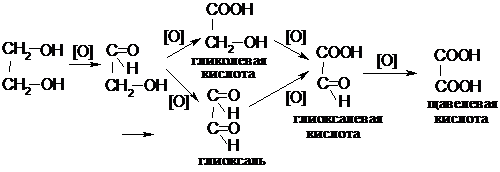
14.2. Двухатомные фенолы.
 |
14.2.1. Номенклатура.
Пирокатехин резорцин гидрохинон
1,2-дигидроксибензен 1,3-дигидроксибензен 1,4-дигидроксибензен
14.2.2. Химические свойства.
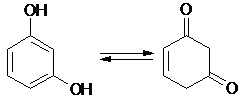 |
Взаимное влияние гидроксильных групп в двухатомных фенолах усиливает их кислотность. Ароматический характер двухатомных фенолов ниже, чем фенола, поэтому для резорцина имеет место кето-енольная таутомерия:
 |
Пирокатехин и гидрохинон легко окисляются, поэтому их применяют как восстановители в фотографии:
о-бензохинон п-бензохинон

