2020-04-20
2020-04-20 96
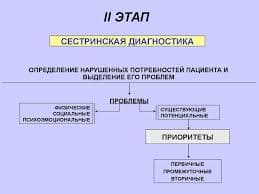
96Конденсація Кневенагеля на сьогоднішній день є загальновідомою та широковживаною в органічному синтезі реакцією формування С-С зв’язку завдяки достатньо широкому колу речовин, що можуть бути використані як субстрати у ній, та різноманітному спектру застосування сполук, що є її продуктами.
На першому етапі досліджень нами була розроблена препаративна методика для проведення цієї реакції. Для цього було оптимізовано розчинник, молярне співвідношення реагентів, час та температуру проведення реакції, що дозволило довести виходи цільових бензиліденових сполук до кількісних. Оптимальними умовами для проведення конденсації Кневенагеля з використанням TMSCl, як конденсуючого та водовіднімаючого агенту на тестовій суміші бензальдегіду 1(1) та малодинітрилу 2(1) (або етилового естеру ціаноцтової кислоти 2(2)) виявилося: нагрівання відповідного альдегіду з метилен активною компонентою у співвідношенні 1:1 при 100 єС в розчині DMF протягом 1-ї години в pressure tube, або при кімнатній температурі протягом 4 годин. Наступним кроком було встановлення меж застосування реакції по метилен активним компонентам та вивчення впливу останніх на умови проведення реакції. Для цього нами були обрані 3 модельні альдегіди: PhCHO (1(1)), p -MeO-C6H4CHO (1(2)), p -Cl-C6H4CHO (1(3)).
При переході від малонодинітрилу 2(1) та етилового естеру ціаноцтової кислоти 2(2) до амідів ціаноцтової кислоти 2(3-10) має місце чітка залежність реакційної здатності від кількості замісників при атомі азоту амідної групи. Так, у разі N- не заміщеного 2(3) та моно N-заміщених 2(4-9) амідів, утворення відповідних продуктів 3{1(1-3)-2(3-9)} відбувається майже з кількісними виходами. Використання N,N-дизаміщених амідів ціаноцтової кислоти 2(10) веде до значного зменшення ступеню конверсії вихідних компонентів в цільовий продукт, а заміна однієї нітрильної групи в малонодинітрилі на карбонільну 2(11-13) або сульфонову 2(14,15) практично не впливає на виходи та не потребує змін в процедурі. (Схема 1)
Наступним кроком дослідження було розширення нашої процедури на отримання бензиліденових похідних гетарилацетонітрилів 4(1-12) та фенілацетонітрилів 4(13,14). (Схема 2)
Поведінка гетарилацетонітрилів виявилася цілком аналогічною до попередніх сполук, а ось фенілацетонітрил 4(13) не реагував з альдегідами ні при кімнатній температурі, ні при багаточасовому нагріванні при 100°С. Збільшивши СН-кислотність фенілацетонітрилу за рахунок введення NO2-групи в бензенове кільце (4(14)), нам вдалося провести конденсацію з альдегідами, однак навіть у цьому випадку конверсія вихідних реагентів не перевищувала 75%.
Продовжуючи пошук границь застосування нашої синтетичної процедури ми дослідили аміди ацетооцтової 6(1,2) та диаміди малонової 8(1-3) кислот. (Схема 3)
Поведінка 6(1,2) подібна до амідів ціаноцтової кислоти 2, однак вони виявилися нестійкими до нагрівання за наших умов, що дозволяло проводити конденсацію лише при кімнатній температурі. При цьому залежність ступеня конверсії від кількості замісників біля атому азоту для 6 повністю збігалася з аналогічною для 2. Перехід до диамідів малонової кислоти 8(1-3) найбільш ярко проілюстрував цю залежність. Так, у випадку сполук 8(1,2), продукти 9{1(1-3)-8(1,2)} були синтезовані нами з високими виходами, а у випадку диаміду піролідину 8(3) 24 годинне нагрівання реакційної суміші за аналогічних умов не приводить до утворення навіть слідових кількостей бензиліденового похідного.
Використання в даній реакції ацилпіруватів 10(1-3) веде до внутрішньо молекулярної циклізації з утворенням продуктів 11. (Схема 4)
Ще одними “класичними” об’єктами в конденсації Кневенагеля є (тіо)барбітурові кислоти 12(1-3) та інші циклічні дикетони 12(4-6) та їх гетероциклічні аналоги 12(7-16). Відомо, що основною проблемою при синтезі бензиліденових похідних такого роду субстратів є можливість конденсації 2-х молекул останніх з однією молекулою альдегіду. Використання системи Me3SiCl/DMF при температурі не вище 25єС дозволило нам селективно отримати бензиліденові похідні 13 для усіх типів субстратів за виключенням циклічних 1,3-дикетонів 12(4,5) та піразолону 12(12), для яких нам так і не вдалося підібрати умови переважного утворення одного з продуктів. (Схема 5)
Циклічні кетони, що містять дві рівноцінні СН-кислотні групи типу 15(1-4) утворюють біс-продукти 16.
При переході від метилен активних сполук, що містять СН-кислотну метиленову групу, до сполук з СН-кислотною метильною групою, типу ацетофенону 17(1) або його гетероциклічних аналогів 17(2,3), а також тетралону 17(4) та його аналогів 17(5,6) необхідно нагрівання протягом 6-10 годин в залежності від природи реагентів. (Схема 6)
Ще більш цікавими, з точки зору створення комбінаторних бібліотек, є метилгетероцикли з СН-кислотною метильною групою, оскільки стирени, що утворюються з них внаслідок реакції, знаходять найбільш широке застосування серед бензиліденових похідних. Слід зауважити, що на сьогоднішній день не існує загальної зручної процедури для синтезу цього класу сполук, що значно скорочує коло субстратів придатних до цього перетворення.
Використовуючи розроблену нами методику, були синтезовані стирени на основі сполук 19(1-19) за допомогою нагрівання реакційної суміші протягом 6-12 годин. Отримання стиренів зі сполук 19(20-32) потребує нагрівання на водяній бані протягом 20-25 годин, однак, внаслідок такого тривалого нагрівання, нам вдалося досягти майже кількісного утворення цільових продуктів. Слід зауважити, що отримання стиренів подібного типу за іншими методиками відбувається з низькими виходами та потребує достатньо жорстких умов, а стирени на основі сполук 19(28-32) взагалі отримані нами вперше. (Схема 7)
З метою краще проілюструвати переваги нашого методу, а також дослідити та висвітлити межі його застосування по карбонільній компоненті нижче наведено невеликий сет з 48 похідних на основі сполуки 19(20) та різноманітних альдегідів 1(1-48). (Схема 8)
Слід зауважити, що реакція утворення бензиліденових похідних за нашим методом відбувається стереоселективно з утворенням виключно транс- ізомеру по відношенню до ймовірного місця силілювання метилен активної сполуки.
Утворення сполук з такою конфігурацією, літературні дані та отримані нами результати наштовхнули на думку про наступну схему перебігу процесу. (Схема 9).
Розширюючи межі застосування нашого підходу в бік використання “нетрадиційних” субстратів в якості метилен активних сполук ми спробували ввести в реакцію Кневенагеля 2-оксиметилбензтіазол 21(1). Виявилося, що ця сполука досить легко реагувала з альдегідами 1 з отриманням відповідних бензтіазолілбензилкетонів 22 з майже кількісними виходами. (Схема 10)
Отримавши такі оптимістичні результати ми вирішили спробувати в цій реакції інші 2-оксиметилгетероцикли 21(2-9). (Схема 11)
При переході до найближчих аналогів 21(2,3) очікувані гетарилбензилкетони 22 утворювалися у кількостях 21% та 44% відповідно, а основними продуктами виявилися гетарилхлорвінільні похідні 23. Аналогічна ситуація була і зі сполуками 21(5,7-9), причому, в разі 21(5), 2-хінолілбензилкетон 22{1(1)-21(5)} виявився основним продуктом реакції (81%), що дозволило отримати цю сполуку в чистому вигляді. В разі використання 21(6) в реакційній суміші окрім продуктів типу 22 та 23 ми фіксували утворення сполуки 24, що є продуктом конденсації 22 з іще однією молекулою альдегіду.
Ці результати дозволили нам ввести в конденсацію Кневенагеля цілий ряд відповідних хлорметильних похідних гетероциклів, взаємодія яких з альдегідами приводила до утворення виключно сполук типу 24. Також була розроблена методика синтезу хлорвінільних похідних 24 виходячи з тозилатів відповідних 2-оксиметиленпохідних 27, вивчена можливість використання TMSBr та TMSI в цій реакції, а також використання F замість OTos в якості нуклеофугу. (Схема 12)
Слід зауважити, що дана реакція також відбувається стереоселективно з утворенням транс- продуктів.