2020-06-12
2020-06-12 140
140Мекониевая непроходимость
Первые симптомы заболевания появляются в большинстве случаев в течение первого года жизни. У30—40%больных муковисцидоз диагностирован впервые дни жизни в виде мекониевой непроходимости. Данная форма заболевания обусловлена отсутствием трипсина, что приводит к скоплению в петлях тонкого кишечника(чаще всего в илеоцекальной области) плотного, вязкого по консистенции мекония. У здорового новорожденного первородный кал отходит на первые, реже — вторые сутки после рождения. У больного ребенка отсутствует выделение мекония. Ко второму дню жизни ребенок становится беспокойным, живот вздут, отмечаются
срыгивания и рвота с примесью желчи. Через 1—2 дня состояние новорожденного ухудшается: кожные покровы сухие и бледные, на коже живота появляется выраженный сосудистый рисунок, тургор тканей снижен, беспокойство сменяется вялостью и адинамией, нарастают симптомы интоксикации и эксикоза.
 Лёгочная (респираторная) форма
Лёгочная (респираторная) форма
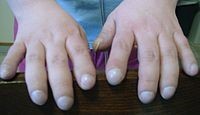
Первыми симптомами бронхо- лёгочной формы муковисцидоза являются вялость, бледность кожных покровов, недостаточная прибавка массы тела при удовлетворительном аппетите. В некоторых случаях (тяжёлое течение) с первых дней жизни у больного появляется покашливание, которое постепенно усиливается и приобретает коклюшеподобный характер. Кашель сопровождается отделением густой мокроты, которая при наслоении бактериальной флоры становится впоследствии слизисто-гнойной. Повышенная вязкость бронхиального секрета приводит к развитию мукостаза и закупорке мелких бронхов и бронхиол, что способствует развитию эмфиземы, а при полной закупорке бронхов формированию ателектазов. У детей раннего возраста в патологический процесс быстро вовлекается паренхима лёгкого, что приводит к развитию тяжёлой, затяжной пневмонии со склонностью к абсцедированию. Поражение лёгких всегда двустороннее. При объективном обследовании отмечаются влажные мелко-и среднепузырчатые хрипы, а при перкуссии выслушивается коробочный оттенок звука. У больных может развиться токсикоз и даже клиника шока на фоне заболеваний, протекающих с высокой температурой тела, или в жаркое время года при значительной потере натрия и хлора с потом. В дальнейшем пневмония приобретает хроническое течение, формируются пневмосклероз и бронхоэктазы, появляются симптомы «лёгочного сердца», лёгочная и сердечная недостаточность. Симптом барабанных палочек и часовых стекол при муковисцидозе.
Кишечная форма
Клиническая симптоматика кишечной формы обусловлена секреторной недостаточностью желудочно-кишечного тракта. Нарушение ферментативной активности желудочно-кишечного тракта особенно ярко выражено после перевода ребенка на искусственное вскармливание или прикорм и проявляется недостаточным расщеплением и всасыванием белков, жиров и в меньшей степени углеводов. В кишечнике преобладают гнилостные процессы, сопровождающиеся накоплением газов, что приводит к вздутию живота. Дефекации частые, отмечается полифекалия (суточный объём каловых масс в 2—8 раз может превышать возрастную норму). После того, как больного муковисцидозом ребенка начинают высаживать на горшок, нередко отмечается выпадение прямой кишки (у 10—20 % больных). Больные предъявляют жалобы на сухость во рту, что обусловлено высокой вязкостью слюны. Больные с трудом пережевывают сухую пищу, а во время еды употребляют значительное количество жидкости. Аппетит в первые месяцы сохранен или даже повышен, но вследствие нарушения процессов пищеварения у больных быстро развивается гипотрофия, полигиповитаминоз. Мышечный тонус и тургор тканей снижен. Больные предъявляют жалобы на боли в животе различного характера: схваткообразные — при метеоризме, мышечные — после приступа кашля, боли в правом подреберье — при наличии правожелудочковой недостаточности, боли в эпигастральной области обусловлены недостаточной нейтрализацией желудочного сока в двенадцатиперстной кишке при сниженной секреции поджелудочной железой бикарбонатов.
Смешанная форма
Смешанная форма муковисцидоза является наиболее тяжёлой и включает клинические симптомы как лёгочной, так и кишечной форм. Тяжесть течения принято оценивать в большинстве случаев характером и степенью поражения бронхо-лёгочной системы.
Различают 4 стадии патологических изменений бронхо-лёгочной системы при муковисцидозе:
· 1-стадия — стадия непостоянных функциональных изменений, которая характеризуется сухим кашлем без мокроты, незначительной или умеренной одышкой при физических нагрузках. Продолжительность данной стадии может составлять до 10лет.
· 2-стадия — стадия развития хронического бронхита, которая характеризуется наличием кашля с отделением мокроты, умеренной одышкой (усиливается при напряжении), формированием деформацией концевых фаланг пальцев. При аускультации выслушиваются влажные, «трескучие» хрипы на фоне жесткого дыхания. Продолжительность данной стадии может составлять от 2 до 15 лет.
· 3-стадия—стадия прогрессирования бронхолёгочного процесса с развитием осложнений. Формируются зоны диффузного пневмофиброза и ограниченного пневмосклероза, бронхоэктазы, кисты и выраженная дыхательная недостаточность в сочетании с сердечной недостаточностью по правожелудочковому типу («лёгочное сердце»). Продолжительность стадии от 3 до 5лет.
· 4-стадия характеризуется тяжёлой кардио-респираторной недостаточностью, которая в течение нескольких месяцев приводит к смерти больного.
Диагноз
Диагноз муковисцидоза определяется данными клинических и лабораторных методов обследования пациента. Для постановки диагноза заболевания необходимо наличие четырёх основных критериев: хронический бронхолёгочный процесс и кишечный синдром, случаи муковисцидоза у сибсов, положительные результаты потового теста. Пот для исследования собирают после электрофореза с пилокарпином. Особое место в диагностике занимает молекулярно-генетическое тестирование. В настоящее время в России по наличию известных мутаций доступны идентификации 65—75 % больных муковисцидозом, что не дает возможности использовать для верификации диагноза заболевания только молекулярно- генетическое обследование.
 Лечение муковисцидоза симптоматическое. Важное значение имеет питание больного. Суточный калораж должен на 10—30 % превышать возрастную норму за счет увеличения в рационе белкового компонента. Потребность в белке удовлетворяют употреблением в пищу мяса, рыбы, яиц, творога. Потребление жиров значительно ограничивают. Можно использовать жиры, в состав которых входят жирные кислоты со средним размером цепи, так как их усвоение не зависит от активности липазы поджелудочной железы.
Лечение муковисцидоза симптоматическое. Важное значение имеет питание больного. Суточный калораж должен на 10—30 % превышать возрастную норму за счет увеличения в рационе белкового компонента. Потребность в белке удовлетворяют употреблением в пищу мяса, рыбы, яиц, творога. Потребление жиров значительно ограничивают. Можно использовать жиры, в состав которых входят жирные кислоты со средним размером цепи, так как их усвоение не зависит от активности липазы поджелудочной железы.
Фенилкетонурия
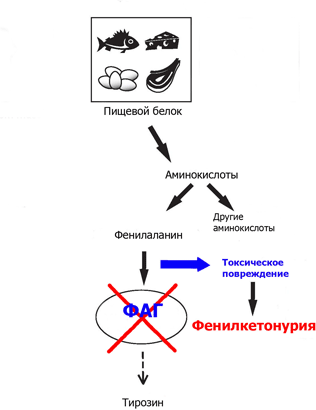
Фенилкетонурия (фенилпировиноградная олигофрения)— наследственное заболевание группы ферментопатий, связанное с нарушением метаболизма аминокислот, главным образом фенилаланина. Сопровождается накоплением фенилаланина и его токсических продуктов, что приводит к тяжёлому поражению ЦНС, проявляющемуся в виде нарушения умственного развития.
История. Открытие фенилкетонурии связывают с именем норвежского врача Ивара Асбьёрна Фёлинга, описавшего в 1934 году гиперфенилаланинемию, ассоциированную с задержкой умственного развития. В Норвегии заболевание известно под названием болезни Фёлинга в честь открывателя.
Этиология. В большинстве случаев (классическая форма) заболевание связано с резким снижением или полным отсутствием активности печёночного фермента фенилаланин-4-гидроксилазы, который в норме катализирует превращение фенилаланина в тирозин. Заболевание наследуется аутосомно-рецессивно и вызвано мутацией гена, локализующегося в длинном плече 12 хромосомы.
 Патогенез
Патогенез
Вследствие метаболического блока активируются побочные пути обмена фенилаланина, и в организме происходит накопление его токсичных производных —
фенилпировиноградной и фенилмолочной кислот, которые в норме практически не образуются. Кроме того, образуются также почти полностью отсутствующие в норме — фенилэтиламини ортофенилацетат, избыток которых вызывает нарушение метаболизма липидов в головном мозге. Это ведёт к прогрессирующему снижению интеллекта у таких больных вплоть до идиотии.
Диагностика
Производится полуколичественным тестом, или количественным определением фенилаланина в крови. При не леченных случаях возможно выявление продуктов распада фенилаланина (фенилкетонов) в моче (не ранее 10-12 дня жизни ребенка).
Лечение и профилактика
При своевременной диагностике патологических изменений можно полностью избежать, если с рождения и до полового созревания ограничить поступление в организм фенилаланина с пищей. Позднее начало лечения хотя и даёт определённый эффект, но не устраняет развившихся ранее необратимых изменений ткани мозга. При рождении ребёнка в роддомах на 3-4 сутки берут анализ крови и проводят неонатальный скрининг для обнаружения врожденных заболеваний обмена веществ. На этом этапе возможно обнаружение фенилкетонурии, и, как следствие, возможно раннее начало лечения для предотвращения необратимых последствий. Лечение проводится в виде строгой диеты от обнаружения заболевания как минимум до полового созревания. Диета исключает мясные, рыбные, молочные продукты и другие продукты содержащие животный и, частично, растительный белок. Дефицит белка восполняется аминокислотными смесями без фенилаланина. Некоторые (мягкие) формы заболевания поддаются лечению ко фактором (тетрагидробиоптерином) пораженного фермента (фенилаланингидроксилазы).
2.3.Адреногенитальный синдром- (врожденная дисфункция коры надпочечников, врожденная гиперплазия коры надпочечников) — группа наследственных болезней, в основе которых лежит недостаточность ферментов на различных уровнях синтеза стероидных гормонов коры надпочечников — кортизона и альдостерона. Тип наследования аутосомно- рецессивный. Частота 1: 5000-1: 6500.
Патогенез. Наследственный дефект в ферментативных системах (в большинстве случаев дефицит или недостаточность 21-гидроксилазы и дефицит 11- гидроксилазы) приводит к снижению содержания в крови кортизона и альдостерона. Синтез половых гормонов при этом в коре надпочечников не нарушается. Низкий уровень кортизола в крови по принципу обратной связи стимулирует гипоталамо- гипофизарную систему и повышение секреции АКТГ. В свою очередь высокий уровень АКТГ способствует гиперплазии коры надпочечников именно той зоны, в которой не нарушен синтез гормонов — преимущественно андрогенов. Одновременно с андрогенами образуются промежуточные продукты синтеза кортизона.
В зависимости от характера ферментативного дефекта выделяют следующие формы адреногенитального синдрома: вирильную (простую, компенсированную) и сольтеряющую.
Вирильная форма — наиболее частая форма синдрома; она обусловлена частичной недостаточностью 21-гидроксилазы. При этой форме нарушается только синтез глюкокортикоидов, что частично компенсируется гиперплазией надпочечников и приводит к латентной надпочечниковой недостаточности. Гиперпродукция андрогенов, начинающаяся еще внутриутробно, приводит к андрогенизации вторичных половых признаков плода и рождению девочек с признаками ложного женского гермафродитизма, а мальчиков — с увеличенным половым членом. Имеет место гиперпигментация наружных половых органов, перианальной зоны, кожных складок, около сосковых кружков молочных желез. Если диагноз после рождения не был поставлен и не проводилось лечение, то в дальнейшем (в среднем в 2-4года)появляются признаки преждевременного полового созревания по мужскому типу: половое оволосение, низкий голос, acnevulgaris, ускорение роста и др. Вследствие раннего закрытия зон роста дети остаются низкорослыми. Степень выраженности указанных симптомов может варьировать в довольно широких пределах.
Диагноз основывается на данных анамнеза и клиники, результатах рентгенографии кистей рук (ускорение костного возраста), выявлении повышенной экскреции с мочой 17-кетостероидов (17-КС), снижения экскреции 17- оксикортикостероидов, высокого уровня в крови АКТГ, 17-оксипрогестерона.
Лечение. Глюкокортикоиды пожизненно. Дозу подбирают индивидуально под контролем содержания 17-КС в суточной моче. Психотерапия. При необходимости проводят оперативную коррекцию наружных половых органов в соответствии с биологическим полом, например, пластику влагалища, клиторэктомию. В ряде случаев решается вопрос о перемене пола.
Прогноз при своевременно начатом лечении для жизни благоприятный.
Сольтеряющая форма (более редкая, обусловлена полным блоком 21- гидроксилазы). При этой форме нарушается синтез не только глюкокортикоидов (гидрокортизона, кортизона), но и минералокортикоидов (альдостерона), что ведет, помимо андрогенизации, к усиленному выводу из организма натрия и хлоридов и к гиперкалиемии. Наиболее ранними симптомами, кроме андрогенизации, являются отмечающиеся с рождения рвота фонтаном, как правило, не связанная с приемом пищи, жидкий стул. Развивается эксикоз, возможны судороги. Прогрессирующее нарушение водно-солевого баланса заканчивается коллапсом и расстройством сердечного ритма, а затем наступает летальный, исход. Клиническая картина пр и этой форме напоминает пилоростеноз (псевдопилоростеноз).
Диагноз основывается на тех же критериях, что и при вирильной форме.
Дифференциальный диагноз, помимо заболеваний, указанных при вирильной форме, проводится с пилоростенозом, кишечными инфекциями, токсическим синдромом.
Лечение. Используют глюкокортикоиды, как при вирильной форме, но в сочетании с минералокортикоидами (дезоксикортикостерона ацетат —ДОКСА).
Прогноз при своевременно начатом лечении относительно благоприятный.
Гипертоническая форма — наиболее редкая, обусловлена дефицитом 11- гидроксилазы, в результате чего, как и при вирильной форме, снижается синтез кортизона и увеличивается продукция андрогенов. По пути синтеза минералокортикоидов снижается образование альдостерона, но в повышенных количествах накапливается 11-дезоксикортикостерон (у здоровых расщепляющийся 11-гидроксилазой). Он обладает минералокортикоидными свойствами и способствует задержке натрия в организме, что обусловливает длительную артериальную гипертензию, осложняющуюся кровоизлияниями в мозг с развитием гемипареза, декомпенсацией сердечной деятельности, изменение глазного дна, сосудов почек и др. Манифестация процесса наступает после 3 лет, но бывает и более раннее начало.
Диагностика и дифференциальная диагностика те же, что и при вирильиой форме, но с учетом артериальной гипертензии.
Лечение то же, что и при вирильной форме. Прогноз для жизни при своевременно начатом лечении благоприятный. Терапия кортикостероидами носит характер заместительной и обеспечивает нормальное развитие ребенка.
Профилактика — медико-генетическое консультирование.
2.4.Галактоземия
Гала́ктоземи́я- наследственное заболевание, в основе которого лежит нарушение обмена веществ на пути преобразования галактозы в глюкозу (мутация структурного гена, ответственного за синтез фермента галактозо-1- фосфатуридилтрансферазы).
Этиология и патогенез
Галактоза, поступающая с пищей в составе молочного сахара — лактозы, подвергается превращению, но реакция превращения не завершается в связи с наследственным дефектом ключевого фермента. Галактоза и ее производная накапливаются в крови и тканях, оказывая токсическое действие на центральную нервную систему, печень и хрусталик глаза, что определяет клинические проявления болезни. Тип наследования галактоземии аутосомно-рецессивный.
Клиника: Заболевание проявляется в первые дни и недели жизни выраженной желтухой, увеличением печени, неврологической симптоматикой (судороги, нистагм (непроизвольное движение глазных яблок), гипотония мышц), рвотой; в дальнейшем обнаруживается отставание в физическом и нервно-психическом развитии, возникает катаракта. Тяжесть заболевания может значительно варьировать; иногда единственным проявлением галактоземии бывают лишь катаракта или непереносимость молока. Один из вариантов болезни — форма Дюарте — протекает бессимптомно, хотя отмечена склонность таких лиц к хроническим заболеваниям печени. При лабораторном исследовании в крови определяется галактоза, содержание которой может достигать 0,8 г/л; специальными методами (хроматография) удается обнаружить галактозу в моче. Активность ферментов в эритроцитах резко снижена или не определяется, содержание ферментов увеличено в 10—20 раз по сравнению с нормой. При наличии желтухи нарастает содержание как прямого (диптюкуронида), так и непрямого (свободного) билирубина. Характерны и другие биохимические признаки поражения печени (гипопротеинемия, гипоальбуминемия, положительные пробы на нарушение коллоидоустойчивости белков). Значительно снижается сопротивляемость по отношению к инфекции. Возможно проявиться и геморагический диатез из-за уменьшения протеино-синтетической функции печени и уменьшение числа тромбоцитов, петехии.
Диагностика и дифдиагностика
Позитивные пробы на сахар и обнаружение галактозы в моче в первые дни жизни, а также уровень ее в крови более 0,2 г/л требуют специального обследования ребенка на галактоземию. Существуют специальные методы определения активности ферментов, превращающихся в галактозу, которые выполняются в централизованных биохимических лабораториях.
Дифференциальный диагноз проводится обычно с сахарным диабетом.
Тяжелые формы заканчиваются летально в первые месяцы жизни, при затяжном течении на первый план могут выступать явления хронической недостаточности печени или поражения центральной нервной системы.
Лечение и профилактика
При подтверждении диагноза необходим перевод ребенка на питание с исключением, главным образом, женского молока. Для этого разработаны специальные продукты: сояваль, нутрамиген, безлактозный энпит. Рекомендуются заменные переливания крови, дробные гемотрансфузии, вливания плазмы. Из лекарственных препаратов показано назначение оротата калия, АТФ, кокарбоксилазы, комплекс витаминов.
С возрастом наблюдается ослабление этого специфического нарушения обмена.
Болезнь Тея-Сакса
Самая распространенная из болезней обмена липидов, болезнь Тея-Сакса вызывается врожденной недостаточностью фермента (гексозаминидазы А). Она характеризуется умственной отсталостью и нарушениями моторики, которые обычно прогрессируют и приводят к гибели ребенка. Дети с болезнью Тея-Сакса чаще всего не доживают до 5лет.
Симптомы
Новорожденные с болезнью Тея-Сакса появляются на свет внешне здоровыми, хотя у них может быть повышение рефлекса Моро, обычно появляющееся в 3- 4 месяца.
В возрасте 3-6 месяцев дети становятся апатичными и реагируют только на громкий звук. Мышцы шеи, туловища, рук и ног слабеют, вскоре ребенок не может садиться и поднимать голову. Ему трудно перевернуться, он не может удерживать предметы, у него нарастает ухудшение зрения.
К 18 месяцам ребенок обычно полностью теряет слух и зрение, у него появляются судороги, генерализованные параличи, спастические движения. Далее клиническая картина только ухудшается из-за повторных бронхопневмоний.