2020-06-08
2020-06-08 174
174Наличие в эритроцитах людей аномальных или патологических гемоглобинов определяет состояния, обозначаемые как гемоглобинозы, или гемоглобинопатии.
Это наследственные аномалии кроветворения, при которых молекулы патологических гемоглобинов имеют измененную структуру, поэтому подобные заболевания относятся к группе так называемых молекулярных болезней. Аномальные гемоглобины различаются своими физико - химическими свойствами, а также по молекулярной структуре глобиновой части.
Гемоглобин S
Отличается от гемоглобина А строением четвертого пептида, в котором на шестом месте вместо глутаминовой кислоты находится электрически нейтральный валин. Гемоглобин S менее растворим, нейтрален по заряду, электрофоретически менее подвижен. В капиллярах при отдаче кислорода гемоглобин S выпадает в осадок в форме веретенообразных кристаллоидов (тактоидов), которые растягивают оболочку и ведут к распаду эритроцитов. У гетерозиготов содержание гемоглобина S равняется 20 - 45 %, у гомозиготов - 60 - 90 %. Гетерозиготная форма аномалии протекает бессимптомно или сопровождается легкой гемолитической анемией. У гомозиготных особей уже с первых месяцев жизни развивается тяжелая форма серповидноклеточной анемии.
Гемоглобин F
Характерный для крови плода фетальный гемоглобин может быть обнаружен в повышенных количествах в эритроцитах крови недоношенных детей, при коклюше, серповидноклеточной анемии, талассемии, врожденной микроцитарной анемии, пернициозной анемии, острых и хронических лейкозах, миеломной болезни. Наибольшее содержание (до 97 %) наблюдается при большой талассемии.
Гемоглобин С
Отличается строением четвертого пептида молекулы гемоглобина, в котором на шестом месте вместо глутаминовой кислоты находится лизин. Центр распространения гена С - северная часть
Гемоглобин D
Обнаружен у 2 % берберов Марокко и у 0,4 % негров США. У гомозиготов наблюдается микроцитоз, слабый анизо- и пойкилоцитоз и мишеневидность эритроцитов. Описано несколько гемоглобинов D (в северо-западной Индии, среди сикхов в Индии, на острове Кипр, в Турции).
Гемоглобин Е
Обнаружен у жителей Юго-Восточной Азии: в Кампучии, Таиланде, Бирме, Бенгалии, у веддов Шри-Ланки, в северо-восточной Малайе, у населения Калимантана и Сулавеси. Частота распространения гена С в разных местностях колеблется от 1 - 3 до 13 (Таиланд) - 20 (Бирма) - 28 - 37 % (Кампучия). У гомозиготов ЕЕ наблюдается микроцитоз, компенсируемый развитием эритроцитоза (до 7 - 8 x 1012 /л).
Гемоглобинопатии
Серповидноклеточная анемия - тяжёлое наследственное заболевание, обусловленное точечной мутацией гена, кодирующего структуру β-цепи гемоглобина. В результате в эритроцитах больных присутствует HbS, β-цепи которого в шестом положении вместо гидрофильной глутаминовой кислоты содержат гидрофобную аминокислоту валин. Появление гидрофобной аминокислоты недалеко от начала молекулы способствует возникновению нового центра связывания, поэтому при низком парциальном давлении кислорода тетрамеры дезокси-HbS ассоциируют, образуя длинные микротрубчатые образования, которые полимеризуются внутри эритроцитов. Полимеризация приводит к нарушению структуры эритроцитов, они приобретают серповидную форму и легко разрушаются. При этом заболевании отмечают анемию, прогрессирующую слабость, отставание в развитии и желтуху.

Талассемии - наследственные заболевания, обусловленные отсутствием или снижением скорости синтеза α- или β-цепей гемоглобина. В результате несбалансированного образования глобиновых цепей образуются тетрамеры гемоглобина, состоящие из одинаковых протомеров. Это приводит к нарушению основной функции гемоглобина - транспорту кислорода к тканям. Нарушение эритропоэза и ускоренный гемолиз эритроцитов и клеток-предшественников при талассемиях приводит к анемии.

При β-талассемии не синтезируются β-цепи гемоглобина. Это вызывает образование нестабильных тетрамеров, содержащих только α-цепи. При этом заболевании в костном мозге из-за преципитации нестабильных α-цепей усиливается разрушение эритробластов, а ускорение разрушения эритроцитов в циркулирующей крови приводит к внутрисосудистому гемолизу. Как известно, для образования фетального гемоглобина р-цепи не требуются (см. раздел 4), поэтому клинически β-талассемия не проявляется до рождения, после чего происходит переключение синтеза HbF на НBА.
В случае α-талассемии недостаток образования α-глобиновых цепей приводит к нарушению образования HbF у плода. Избыточные γ-цепи образуют тетрамеры, называемые гемоглобином Барта. Этот гемоглобин при физиологических условиях имеет повышенное сродство к кислороду и не проявляет кооперативных взаимодействий между протомерами. В результате гемоглобин Барта не обеспечивает развивающийся плод необходимым количеством кислорода, что приводит к тяжёлой гипоксии. При α-талассемии отмечают высокий процент внутриутробной гибели плода.
Наследственный сфероцитоз. Причиной этой патологии чаще всего является дефект белков цитоскелета эритроцитов - спектрина или ан-кирина, которые обеспечивают поддержание двояковогнутой формы клетки и эластичности мембраны. Эритроциты приобретают шарообразную форму, что приводит к уменьшению площади их поверхности и снижению скорости газообмена. Потеря эластичности клеточной мембраны приводит к повышению хрупкости и травматичности клеток и, как следствие, к ускорению их разрушения в сосудистом русле и селезёнке. Заболевание сопровождается анемией и желтухой. Удаление селезёнки (спленэктомия) при наследственном сфероцитозе улучшает состояние больных, так как предотвращает разрушение сфероцитов в селезёнке.
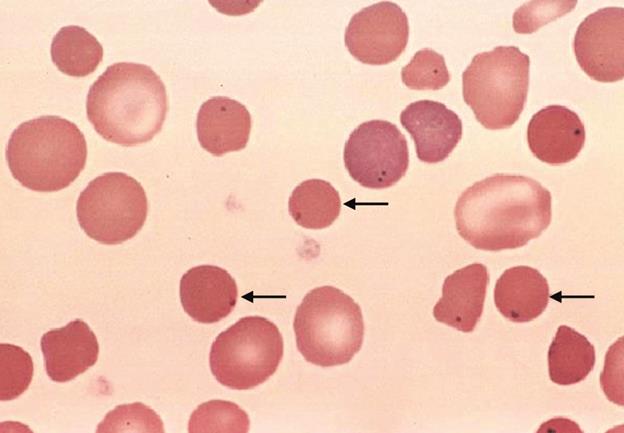
Мегалобластная (макроцитарная) анемия развивается при дефиците фолиевой кислоты или витамина В12.
Фолиевая кислота в виде кофермента (Н4-фолата) участвует в синтезе нуклеотидов. Недостаток фолиевой кислоты приводит к снижению скорости синтеза ДНК в быстроделящихся клетках, и в первую очередь в предшественниках эритроцитов. Клетки дольше пребывают в интерфазе, синтезируя гемоглобин, и становятся крупнее. Кроме того, из-за недостатка нуклеотидов они реже делятся, и количество эритроцитов снижается, а крупные мегалобласты быстрее разрушаются. Всё это в конечном итоге приводит к развитию анемии.
Аналогичная симптоматика развивается при недостатке в организме витамина В12. Этот витамин участвует в переносе метальной группы с N5-метил-Н4-фолата на гомоцистеин с образованием метионина и Н4-фолата (см. раздел 10). Недостаточность витамина В12 приводит к накоплению N5-метил-Н4-фолата в клетках. Дефицит Н4-фолата приводит к нарушению деления клеток и развитию анемии.