2020-06-08
2020-06-08 127
127Реакции 1 - реакция, катализируемая АДГ; 2 - реакция, катализируемая АлДГ; 3 - реакция, катализируемая ацетил-СоА-синтетазой; Скорость образования ацетальдегида (1) при приеме большого количества алкоголя выше, чем скорость его окисления (2), поэтому ацетальдегид накапливается и оказывает влияние на структуру и синтез белков, а также снижает концентрацию восстановленного глутатиона (5), что приводит к активации перекисного окисления липидов (ПОЛ). Скорость глюконеогенеза (6) снижается за счет повышения концентрации НАДН, образующегося при окислении этанола. Лактат (7) выделяется в кровь, что приводит к развитию лактоацидоза. Увеличение концентрации НАДН замедляет скорость ЦТК. В результате происходит накопление ацетил-СоА и активируется синтез кетоновых тел (8). Окисление жирных кислот замедляется (9); увеличивается интенсивность синтеза триглицеридов (10) и холестерина (10), что приводит к ожирению, гипертриацилглицеролемии, и, в конечном счете атеросклерозу, жировому перерождению и циррозу печени.
На начальных стадиях алкоголизма, окисление ацетил-СоА в ЦТК является основным источником энергии в клетке. Избыток ацетил-СоА, образовавшегося из этанола, выводится из митохондрий в виде цитрата и используется в цитоплазме для синтеза жирных кислот и холестерина. Этот процесс требует использования АТФ и НАДФН. Последний образуется в ходе двух реакций окислительной фазы пентозофосфатного пути окисления глюкозы. В этих условиях, снижение скорости глюконеогенеза приводит к дефициту эндогенной глюкозы, синтезированной de novo. Возникающий в связи с этим дефицит НАДФН приводит к дисбалансу в системе работы ферментов антиокислительной защиты (глутатионпероксидаза/глутатионредуктаза). НАДФН является коферметном глутатионредуктазы, осуществляющей поддержку пула восстановленного глутатиона:
GSH + H2O2 GSSG + 2H2O (ГПО)
GSSG + НАДФН+Н+ 2GSH + НАДФ+
С другой стороны, у лиц, страдающих алкогольной зависимостью, нередко наблюдается дефицит тиамина (витамин В1) в организме из-за нарушения режима питания. Кроме того, показано, что алкоголь подавляет всасывание тиамина, поскольку ингибирует кишечную АТФазу, участвующую в абсорбции В1. Поэтому, при введении им глюкозы, может происходить быстрое накопление пирувата и лактата, приводящее к лактоацидозу, нередко со смертельным исходом. С недостаточностью тиамина связывают развитие синдрома Вернике-Корсакова, который обычно включает два состояния:
- тяжелое психическое расстройство (расстройство сознания, паралич глазодвигательных мышц, нарушение координации движений (особенно нижних конечностей, энцефалопатия Вернике).
- хронический нейропсихопатический синдром, характеризующийся поведенческими расстройствами и нарушениями памяти (корсаковский психоз).
Роль витамина В1 в развитии этих нарушений доказана терпевтическим эффектом введения тиамина.
Из жирных кислот и глицерол-3-фосфата образуются триацилглицеролы (ТАГ), которые секретируются в крови в составе ЛПОНП. Повышенная секреция ЛПОНП печенью приводит к развитию гипертриацилглицеролемии. При хроническом алкоголизме, снижение интенсивности синтеза фосфолипидов и белков в печени, в том числе, апобелков, входящих в состав ЛПОНП, вызывает накопление ТАГ в гепатоцитах и жировое перерождение печени.
В период острой алкогольной интоксикации может возникать недостаток оксалоацетата из-за избыточного образования ацетил-СоА. С другой стороны, происходит замедление реакций ЦТК вследствие увеличения содержания НАДН. В этих условиях, избыток ацетил-СоА используется для синтеза кетоновых тел (ацетоацетат и β-гидроксибутират), что может стать причиной возникновения кетоацидоза.
Повышенное образование НАДН и ацетальдегида в ходе окисления этанола приводит к смещению равновесия реакции, катализируемой АДГ в сторону образования этанола. Этанол способен растворяться в липидах клеточных мембран, нарушая при этом их микровязкость и селективные свойства. Это приводит к нарушениям процессов мембранного транспорта, работы ионных каналов, межклеточных контактов и взаимодействия мембранных рецепторов с сигнальными молекулами, что приводит к фатальным сдвигам в ключевых процессах, регулирующих функциональное состояние клеток. Кроме того, этанол способен проходить через клеточные мембраны и проникать в кровь, лимфу и далее в любые ткани организма.
Повреждение печени, наблюдаемое при хроническом употреблении алкоголя, может быть не связано напрямую с воздействием этанола или его метаболитов на гепатоциты. Имеются сведения, что данный процесс может происходить опосредованно, и ключевая роль в нем принадлежит факторам, высвобождаемым в ответ на действие этанола и ацетальдегида. В частности, большое значение приписывается бактериальным эндотоксинам кишечного происхождения. Данные эффекты связывают с повышением проницаемости стенки кишечника и нарушением моторики ЖКТ при воздействии избыточного количества этанола, что способствует стазу и избыточному размножению микрофлоры толстого кишечника, а иногда и колонизации нижних отделов тонкого кишечника. Кроме того, кишечные микроорганизмы способны сами окислять этанол с образованием ацетальдегида, повреждающего кишечный эпителий и увеличивающий проницаемость стенки кишечника для бактериальных токсинов. Медиаторами алкогольиндуцированной гепатотоксичности в данном случае выступают цитокины, в частности фактор некроза опухолей (ФНО-α), секреция которого стимулируется бактериальными эндотоксинами. При этом принципиальное значение имеет совместное воздействие этанола и ФНО-α, так как отдельное воздействие каждого из этих факторов не всегда бывает достаточным для гибели гепатоцитов. Эффекты совместного воздействия этанола и ФНО-α могут реализовываться, по меньшей мере, двумя путями. Во-первых, как уже отмечалось выше, хроническое воздействие этанола и его метаболитов вызывает нарушение функций митохондрий, которые являются основной мишенью, как для этанола, так и для ФНО-α. Это приводит к снижению интенсивности окислительного фосфорилирования и генерации избыточных количеств АФК. В основе второго механизма лежит нарушение работы внутриклеточных защитных систем: антиоксидантной и антиапоптотической. При хроническом воздействии алкоголя резко снижается уровень восстановленного глутатиона в клетках. Кроме того, установлено подавляющее действие алкоголя на индукцию белковых факторов NF-κB и bcl-XL, которые предотвращают гибель клетки после воздействия ФНО-α и входят в систему защиты клетки от апоптоза. Таким образом, гибель гепатоцитов происходит от совместного воздействия этанола и ФНО-α либо путем апоптоза, индуцированного ФНО-α, либо путем некроза, которому способствует снижение запасов АТФ в клетках, вызванное разобщением окислительного фосфорилирования.
Окислительный стресс оказывает выраженное цитотоксическое влияние через трансмембранный рецептор Fas и его лиганд (FasL). У пациентов с алкогольной болезнью печени отмечается высокий уровень FasL в гепатоцитах, обусловленный генерацией АФК. Поскольку на поверхности гепатоцитов экспрессирован также рецептор Fas, это свидетельствует о том, что клетки печени могут быть вовлечены в апоптоз через пара- или аутокринные механизмы (Рис. 10).
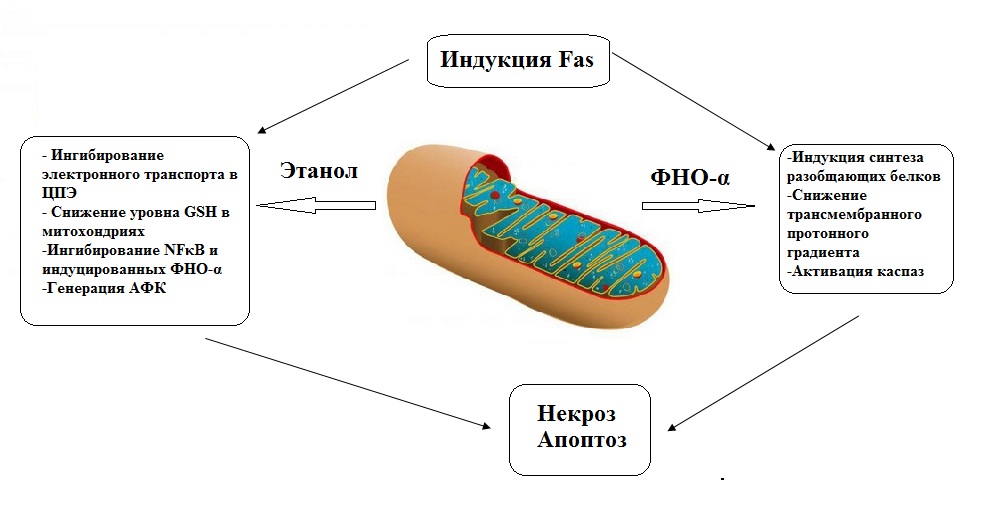
Рис. 10 Механизмы повреждающего действия этанола и ФНО-α на гепатоциты