2020-09-24
2020-09-24 511
511В 1912 году Л.Кляйзен открыл интересную и своеобразную перегруппировку аллиловых эфиров фенолов в аллилфенолы, которая стала прототипом для многих родственных сигматропных перегруппировок. Аллиловый эфир фенола при нагревании до 200-220оС превращается в орто-аллилфенол, т.е. аллильная группа мигрирует в орто-положение бензольного кольца.
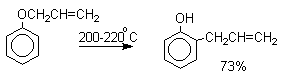
Если оба орто-положения заняты заместителями, то аллильная группа перемещается в пара-положение:
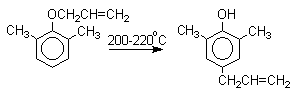
Установлено, что и орто- и пара-перегруппировки являются внутримолекулярными реакциями первого порядка, которые сопровождаются инверсией мигрирующей аллильной группы, т.е. аллильная группа присоединяется к бензольному кольцу своим 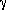 -углеродным атомом.
-углеродным атомом.
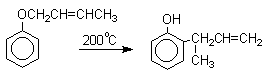
Из этого следует, что переходное состояние перегруппировки Кляйзена должно быть циклическим шестизвенным. Такое переходное состояние включает шесть  -электронов и является ароматическим, что составляет движущую силу этой термической перегруппировки. На последней стадии происходит изомеризация циклогексадиенона в о-аллилфенол. Эта стадия полностью аналогична изомеризации кетона в енольную форму.
-электронов и является ароматическим, что составляет движущую силу этой термической перегруппировки. На последней стадии происходит изомеризация циклогексадиенона в о-аллилфенол. Эта стадия полностью аналогична изомеризации кетона в енольную форму.
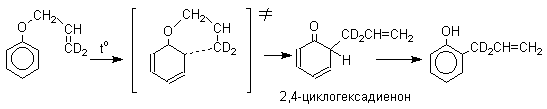
2,4-Циклогексадиенон является интермедиатом перегруппировки аллилариловых эфиров. Такой интермедиат может быть выделен при перегруппировке аллилового эфира 2,6-диметилфенола, когда аллильная группа мигрирует в пара-положение, поскольку енолизация кетона в фенол в этом случае не может происходить из орто-положения. Конечным результатом двух последовательных миграций аллильной группы является сохранение структуры мигрирующей группы.
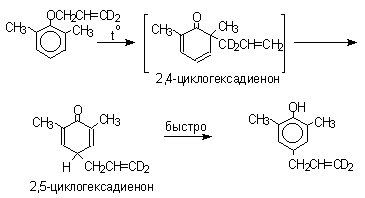
Кроме того, при проведении перегруппировки в присутствии малеинового ангидрида 2,4-циклогексадиенон улавливается в виде аддукта диенового синтеза.
Миграция аллильной группы характерна не только для аллиловых эфиров фенолов. Аллиловые эфиры енолов также подвергаются аллильной перегруппировке. Например, аллилвиниловые эфиры в результате миграции аллильной группы превращаются в , 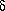 -ненасыщенные карбонильные соединения.
-ненасыщенные карбонильные соединения.
Окисление фенолов
Окисление пространственно незатрудненных фенолов относится к числу сложных, многостадийных процессов, механизм которых мало изучен. Очевидно лишь то, что механизм окисления может сильно меняться в зависимости от природы одно- или двухэлектронного окислителя. Сам фенол при окислении двухэлектронным окислителем - бихроматом натрия или MnO2 в серной кислоте образует с удовлетворительным выходом пара-хинон.
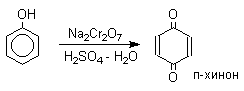
Универсальным одноэлектронным окислителем фенолов является соль Фреми - нитрозодисульфонат калия - редкий пример стабильного неорганического нитроксильного свободного радикала, полученного впервые еще в 1845 году. Окисление фенолов солью Фреми идет в очень мягких условиях по радикальному механизму и приводит к пара-хинонам с выходами, близкими к количественному.
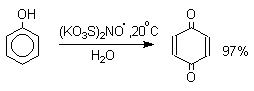
Ароматические амины также гладко окисляются солью Фреми до пара-хинонов.
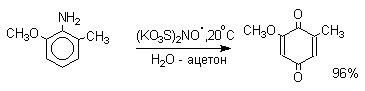
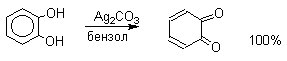
Самый простой и удобный способ получения орто- и пара-бензохинонов состоит в окислении соответственно пирокатехина и гидрохинона. В качестве окислителей можно использовать самые разнообразные реагенты: дихромат натрия, ферроцианид калия, перкислоты, оксид серебра, тетраокись азота и ряд других окислителей.
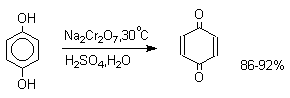
Аналогично ведут себя и пара-аминофенолы.
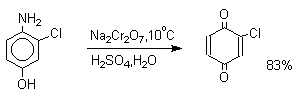
Удовлетворительные результаты при получении орто-бензохинона из пирокатехина достигаются в том случае, если в качестве окислителя используется оксид серебра в эфире в присутствии сульфата натрия для связывания выделяющейся при окислении воды.
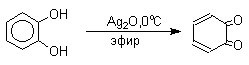
Хиноны, содержащие электроноакцепторные заместители - 2,3,5,6-тетрахлор-1,4-бензохинон (хлоранил) или 2,3,5,6-тетрациано-1,4-бензохинон- служат превосходными окислителями для превращения двух- и трехатомных фенолов в хиноны, например:
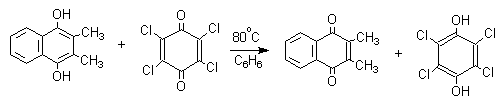
Окисление двухатомных фенолов до 1,4- или 1,2-хинонов осуществляется в несколько стадий, включающих образование анион-радикалов и радикалов. Механизм этой важной в теоретическом отношении модельной реакции будет рассмотрен в разделе "хиноны".
Механизм окисления фенолов, содержащих в обоих орто-положениях и пара-положении алкильные, арильные или алкоксильные группы, тщательно изучен. Е.Мюллер (1953 год) и независимо от него К.Кук обнаружили, что при окислении 2,4,6-три-трет-бутилфенола гексацианоферратом(III) калия K3Fe(CN)6 в бинарной системе бензол-вода в инертной атмосфере образуется устойчивый радикал одновалентного кислорода - три-трет-бутилфеноксил, окрашенный в синий цвет.
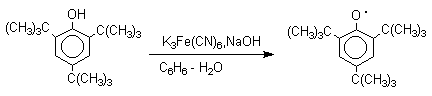
Этот радикал находится в мономерной форме в 0,1 молярном растворе в бензоле или эфире, а также в кристаллическом состоянии. Он очень чувствителен к действию кислорода воздуха, оксида азота (IY), оксида азота (II) и других радикальных частиц. Окисление пространственно затрудненных фенолов до феноксильных (ароксильных) радикалов осуществляется под действием гексацианоферрата (III) калия в бинарной системе бензол-вода, диоксида свинца PbO2, оксида серебра, соли Фреми или другого одноэлектронного окислителя в индифферентной среде, а также электрохимически.
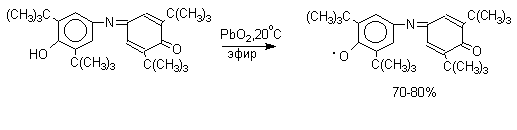
Феноксильные радикалы, содержащие в обоих орто- и пара-положении третичные алкильные группы, мономерны и не проявляют тенденции к образованию димеров в растворе. С другой стороны феноксильные радикалы, содержащие фенильную или алкоксильную группу в орто- или пара-положениях к радикальному центру, проявляют ярко выраженную тенденцию к образованию димеров. Так, например, при окислении 2,4,6-трифенилфенола гексацианоферратом (III) калия получается с выходом 95% димер, который в 0,01 бензольном растворе при 20оС диссоциирован только на 10%. Радикалы с алкоксильной группой в пара-положении более стабильны и 2,6-ди-тре-бутил-4-метоксифеноксил в 3%-ном растворе в бензоле мономерен на 70-75%.
При окислении фенолов образуется несколько различных форм димеров в результате образования новых связей С-С между орто-орто, орто-пара- и пара-пара-положениями исходных радикалов, а также новых С-О связей между атомом кислорода одного радикала и орто-положением другой радикальной частицы. Всего, таким образом, образуется потенциально не менее пяти различных типов димеров, которые находится в равновесии с исходным феноксильным радикалом. Например: для монозамещенного фенола:
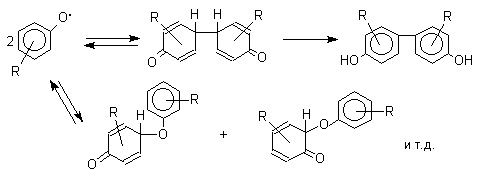
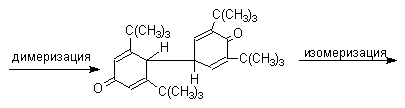
Димеры, возникающие в результате рекомбинации радикалов с образованием новой С-С связи, называются хинолидами. Хинолиды далее изомеризуются с образованием изомерных дигидроксибифенилов. Другой тип димеров, содержащих центральную связь С-О, носит название хиноловых эфиров. Образование хиноловых эфиров свойственно для многих пространственно затрудненных феноксильных радикалов, например, для 2,4,6-трифенилфеноксила; 2,6-ди-трет-бутил-4-фенилфеноксила; 2,6-ди-трет-бутил-4-алкоксифеноксилов и т.д. Хиноловые эфиры могут быть получены прямым окислением соответствующих пространственно затрудненных фенолов.
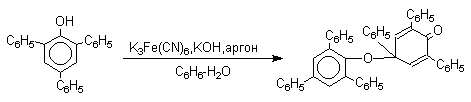
В растворе или при нагревании они диссоциируют на радикалы (см. выше). Хинолидная форма димера образуется в том случае, когда пара-положение или одно из орто-положений феноксила не замещено. Другими словами, хинолиды получаются при окислении 2,4- и 2,6-диалкилфенолов с последующей димеризацией образующихся феноксильных радикалов.
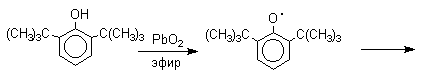
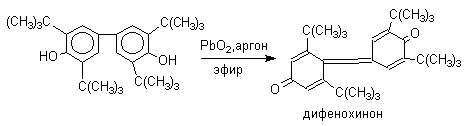
Дальнейшее окисление хинолидного димера, или его таутомерной формы - дигидроксибифенила - приводит к образованию дифенохинона.
Таким образом, производные дифенохинона являются главными конечными продуктами окисления 2,6-диалкилфенолов такими окислителями как: PbO2, Ag2O, K3Fe(CN)6.
Хиноны
Хиноны по своей структуре являются циклогексадиенонами, но их название происходит от ароматических углеводородов: бензохинон от бензола, толухинон от толуола, нафтохинон от нафталина и т.д. Цифры в начале названия обозначают положение двух карбонильных групп.
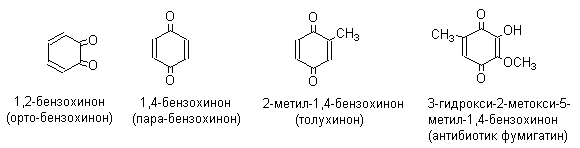
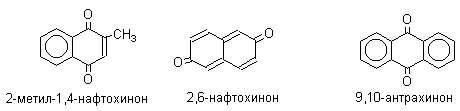
Многие производные хинонов составляют важную группу природных веществ - красителей, пигментов, антибиотиков, витаминов и т.д.
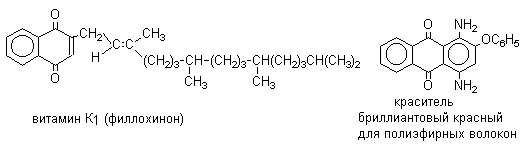
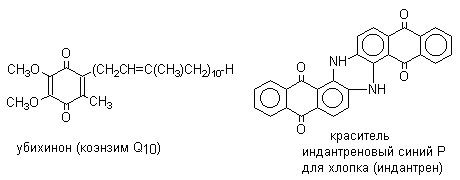
Производные 9,10-антрахинона широко используются в качестве синтетических антрахиноновых красителей для хлопка, шерсти и синтетических волокон. Эти красители отличаются яркостью цвета, высокой термической и фотохимической устойчивостью. Выше в качестве примера приведены формулы некоторых антрахиноновых красителей. Эти красители получают из 2-аминоантрахинона и его производных, например:
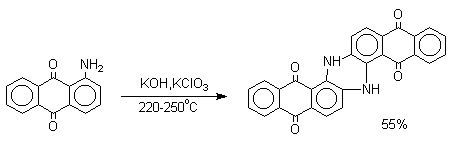
Получение хинонов
Хиноны получают окислением одно- и двухатомных фенолов, аминов и диаминов ароматического ряда. Самый удобный способ получения хинонов заключается в окислении одноатомных фенолов солью Фреми - нитрозодисульфонатом калия. Эта реакция осуществляется в исключительно мягких условиях в водном спирте или ацетоне, выход обычно превышает 90%.
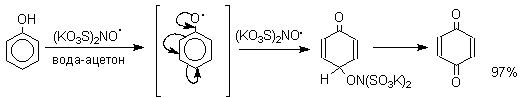
Приведенный на этой схеме циклогексадиеноновый интермедиат был выделен, что доказывает механизм одноэлектронного окисления фенолов солью Фреми. Другим одноэлектронным окислителем фенолов является карбонат серебра. Этот реагент, согласно данным последних лет, особенно пригоден для окисления 1,2-дигидроксибензола и его производных до орто-хинона.
Уникальным реагентом для получения орто-хинонов из одноатомных фенолов оказался (С6Н5SeO)2O.
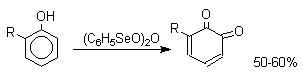
Для окисления фенолов, ароматических аминов и гидрохинонов до 1,4-бензохинонов и 1,4-нафтохинонов часто используют реагенты на основе хрома (YI). К ним относятся оксид хрома (YI) в уксусной кислоте или реагент Килиани (дихромат натрия и серной кислоты), однако выходы хинонов, как правило, ниже, чем при окислении солью Фреми или карбонатом серебра.
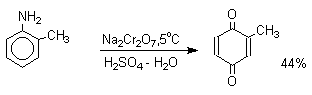
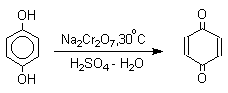
1,4-нафтохинон, 9,10-антрахинон и 9,10-фенантренхинон могут быть получены прямым окислением углеводородов оксидом хрома (YI) в уксусной кислоте или бихроматом натрия в серной кислоте.
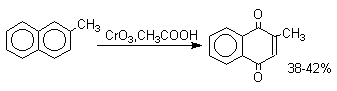
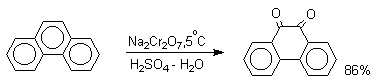
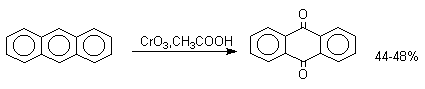
В промышленности тот же самый результат достигается при окислении кислородом в присутствии оксида ванадия (Y) как катализатора. Таким способом можно получать антрахинон и фенантренхинон.
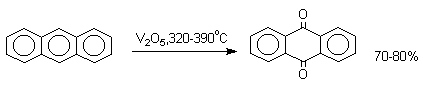
9,10-Антрахинон получают также ацилированием бензола фталевым ангидридом по Фриделю-Крафтсу с последующей циклизацией орто-бензоилбензойной кислоты.
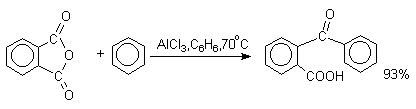
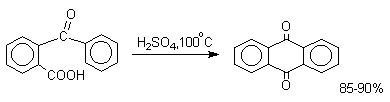
Этот один из самых простых и старых способов промышленного получения антрахинона.