2014-02-17
2014-02-17 2164
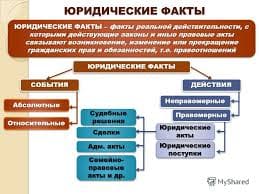
2164Катаболизм ГАГ
Регуляция синтеза ГАГ
Синтез ГАГ
1. На рибосомах синтезируется коровый белок, который по ЭПР поступает в аппарат Гольджи.
2. В аппарате Гольджи с участием трансфераз, локализованных на мембране, на коровом белке путём последовательного присоединения моносахаридов растет цепь ГАГ.
а). Сначала на коровом белке образуется связующий трисахарид: галактоза-галактоза-ксилоза, который может быть присоединен к коровому белку: 1). О-гликозидной связью между серином и ксилозой; 2). О-гликозидной связью между серином или треонином и N-ацетилгалактозамином; 3). N-гликозиламиновой связью между амидным азотом аспарагина и N-ацетилглюкозамином.
б). Затем, на связующем трисахариде наращивается полисахаридная цепь ГАГ. Донорами моносахаридов обычно являются соответствующие УДФ-производные: УДФ-глюкоза, УДФ-глюкуроновая кислота, УДФ-N-ацетилглюкозамин, УДФ-N-ацетилгалактозамин и т.д.
УДФ-глюкуронат образуется при окислении УДФ-глюкозы.
N-ацетилглюкозамин, N-ацетилгалактозамин и сиаловой кислоты синтезируются в соединительной ткани из фруктозо-6-ф (образуется из глюкозы). Источником NH2-группы для аминосахаров служит глутамин. Аминосахар далее ацетилируется с помощью ацетил-КоА. Затем образуются их УДФ-производные.
3. Некоторые углеводы в составе ГАГ сульфируются сульфотрансферазами, донором сульфатной группы выступает ФАФС.
4. L-идуроновая кислота образуется в составе ГАГ в результате реакции эпимеризации D-глюкуроновой кислоты.
Синтез ГАГ стимулируют инсулин, СТГ, половые гормоны.
Синтез ГАГ тормозят глюкокортикоиды.
ГАГ отличаются высокой скоростью обмена: Т½ многих из них составляет от 3 до 10 дней (только для кератансульфата 120 дней).
Разрушение ГАГ начинается в матриксе с участием экзо- и эндогликозидаз и сульфатаз (гиалуронидаз, глюкуронидаз, галактозидаз, идуронидаз и др.).
Затем, из внеклеточного пространства фрагменты ГАГ фагоцитируются клетками и гидролизуются до мономеров с участием лизосомальные гидролаз.
Мукополисахаридозы — заболевания, связанные с генетическим дефектом гидролаз, участвующих в катаболизме ГАГ.
Эти заболевания характеризуются избыточным накоплением ГАГ в тканях, приводящим к деформации скелета и увеличению органов, содержащих большие количества внеклеточного матрикса. Обычно поражаются ткани, в которых в норме синтезируются наибольшие количества ГАГ. В лизосомах при этом накапливаются фрагменты ГАГ, а с мочой выделяются олигосахариды из ГАГ.
Проявляются мукополисахаридозы значительными нарушениями в умственном развитии детей, поражениями сосудов, помутнением роговицы, деформациями скелета, низкой продолжительность жизни.
Эти болезни в настоящее время не поддаются лечению.