 2015-01-30
2015-01-30 2357
2357При комнатной температуре и атмосферном давлении происходит окисление NO кислородом воздуха:
2NO + O2 > 2NO2
Для NO характерны также реакции присоединения галогенов с образованием нитрозилгалогенидов, в этой реакции NO проявляет свойства восстановителя:
2NO + Cl2 > 2NOCl (нитрозилхлорид).
В присутствии более сильных восстановителей NO проявляет окислительные свойства:
2SO2 + 2NO > 2SO3 + N2^.
В воде NO мало растворим и с ней не реагирует, являясь несолеобразующим оксидом.
Нитраты – соли азотной кислоты, являются элементом питания растений и естественным компонентом пищевых продуктов растительного происхождения. Их высокая концентрация в почве абсолютно не токсична для растений, напротив, она способствует усиленному росту надземной части растений, более активному протеканию процесса фотосинтеза, лучшему формированию репродуктивных органов и, следовательно, более высокому урожаю.
Нерациональное применение удобрений, как и несоблюдение других агротехнических требований, обуславливает увеличение остаточного содержания нитратов в растениях. Концентрация нитратов в овощах, зеленых культурах колеблется в широких пределах и может достигать очень больших величин (свекла – 1070 мг/кг, морковь – 180 мг/кг, молодой картофель – 170 мг/кг). В сочетании с нитратами питьевой воды это увеличивает нагрузку загрязнителя на население. Кроме того, нитраты широко используются в различных отраслях промышленности (пищевой, химической, текстильной, резиновой, металлургической) и фармакологии. Таким образом, нитросоединения могут поступать в организм человека с овощами и фруктами, колбасными и консервными изделиями, питьевой водой, вдыхаемым воздухом и лекарствами.
При остром отравлении нитратами у человека возникает метгемоглобиния различной степени тяжести (вплоть до летального исхода); при хроническом отравлении – рак желудка, нарушение работы нервной и сердечно-сосудистой систем. Чувствительность к нитратам повышают факторы кислородного голодания: высокогорье, повышенная концентрация угарного газа, присутствие окислов азота, алкоголь.
Азотные удобрения — неорганические и органические азотосодержащие вещества, которые вносят в почву для повышения урожайности. К минеральным азотным удобрениям относят амидные, аммиачные и нитратные. Азотные удобрения получают главным образом из синтетического аммиака. Из-за высокой мобильности соединений азота, его низкое содержание в почве часто лимитирует развитие культурных растений, поэтому внесение азотных удобрений вызывает большой положительный эффект.
Из всех типов удобрений азотные наиболее подвержены воздействию со стороны почвенных микроорганизмов. В первую неделю после внесения до 70 % массы удобрения потребляется бактериями и грибами (иммобилизуются), лишь после их гибели входящий в их состав азот может использоваться растениями. Большие потери азота удобрений происходят из-за выноса легкорастворимых нитратов и солей аммония из почвенного профиля, а также в ходе денитрификации (газообразные потери) и из-за нитрификации (образование нитратов и их вынос). В итоге коэффициент использования удобрений растениями редко достигает 50 %, их применение может вызывать эвтрофикацию близлежащих водоёмов. Образующийся в ходе денитрификации N2O является сильным парниковым газом.
Гидриды ЭН3 элементов VA подгруппы. Строение молекул. Аммиак. Получе-ние. Термодинамические характеристики реакции синтеза аммиака. Аммиакаты. Гидразин и соли гидрозония. Гидроксиламин и соли гидроксиламмония. Азотистоводородная кислота и азиды.
Аммиа́к — NH3, нитрид водорода, при нормальных условиях — бесцветный газ с резким характерным запахом (запах нашатырного спирта).
Плотность аммиака почти вдвое меньше, чем у воздуха. Молекула аммиака имеет форму тригональной пирамиды с атомом азота в вершине. Три неспаренных p-электрона атома азота участвуют в образовании полярных ковалентных связей с 1s-электронами трёх атомов водорода (связи N−H), четвёртая пара внешних электронов является неподелённой, она может образовать ковалентную связь по донорно-акцепторному механизму с ионом водорода, образуя ион аммония NH4+. Благодаря тому, что не связывающее двухэлектронное облако строго ориентировано в пространстве, молекула аммиака обладает высокой полярностью, что приводит к его хорошей растворимости в воде.
В жидком аммиаке молекулы связаны между собой водородными связями. Сравнение физических свойств жидкого аммиака с водой показывает, что аммиак имеет более низкие температуры кипения (tкип −33,35 °C) и плавления (tпл −77,70 °C), а также более низкую плотность, вязкость (вязкость жидкого аммиака в 7 раз меньше вязкости воды), проводимость и диэлектрическую проницаемость. Это в некоторой степени объясняется тем, что прочность этих связей в жидком аммиаке существенно ниже, чем у воды, а также тем, что в молекуле аммиака имеется лишь одна пара неподелённых электронов, в отличие от двух пар в молекуле воды, что не дает возможность образовывать разветвлённую сеть водородных связей между несколькими молекулами. Аммиак легко переходит в бесцветную жидкость с плотностью 681,4 кг/м³, сильно преломляющую свет. Подобно воде, жидкий аммиак сильно ассоциирован, главным образом за счёт образования водородных связей. Жидкий аммиак практически не проводит электрический ток. Жидкий аммиак — хороший растворитель для очень большого числа органических, а также для многих неорганических соединений. Твёрдый аммиак — бесцветные кубические кристаллы.
Химические свойства.. Благодаря наличию неподеленной электронной пары во многих реакциях аммиак выступает как основание Бренстеда или комплексообразователь (не следует путать понятия «нуклеофил» и «основание Бренстеда». Нуклеофильность определяется сродством к положительно заряженной частице. Основание имеет сродство к протону. Понятие «основание» является частным случаем понятия «нуклеофил»). Так, он присоединяет протон, образуя ион аммония:
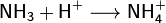
Водный раствор аммиака («нашатырный спирт») имеет слабощелочную реакцию из-за протекания процесса:
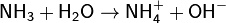
Взаимодействуя с кислотами даёт соответствующие соли аммония:
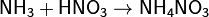
Аммиак также является очень слабой кислотой (в 10 000 000 000 раз более слабой, чем вода), способен образовывать с металлами соли — амиды. Соединения, содержащие ионы NH2−, называются амидами, а N3− — нитридами. Амиды щелочных металлов получают, действуя на них аммиаком:
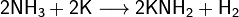
Амиды, имиды и нитриды ряда металлов образуются в результате некоторых реакций в среде жидкого аммиака. Нитриды можно получить нагреванием металлов в атмосфере азота.
Амиды металлов являются аналогами гидроксидов. Эта аналогия усиливается тем, что ионы ОН− и NH2−, а также молекулы Н2O и NH3 изоэлектронны. Амиды являются более сильными основаниями, чем гидроксиды, а следовательно, подвергаются в водных растворах необратимому гидролизу:
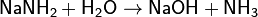
и в спиртах:
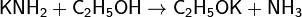
Подобно водным растворам щелочей, аммиачные растворы амидов хорошо проводят электрический ток, что обусловлено диссоциацией:
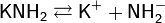
Фенолфталеин в этих растворах окрашивается в малиновый цвет, при добавлении кислот происходит их нейтрализация. Растворимость амидов изменяется в такой же последовательности, что и растворимость гидроксидов: LiNH2 — нерастворим, NaNH2 — малорастворим, KNH2, RbNH2 и CsNH2 — хорошо растворимы.
При нагревании аммиак разлагается, проявляет восстановительные свойства. Так, он горит в атмосфере кислорода, образуя воду и азот. Окисление аммиака воздухом на платиновом катализаторе даёт оксиды азота, что используется в промышленности для получения азотной кислоты:
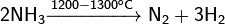 (реакция обратима)
(реакция обратима)
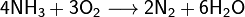
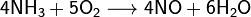
На восстановительной способности NH3 основано применение нашатыря NH4Cl для очистки поверхности металла от оксидов при их пайке:
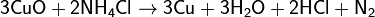
Окисляя аммиак гипохлоритом натрия в присутствии желатина, получают гидразин:
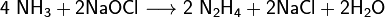
Галогены (хлор, йод) образуют с аммиаком опасные взрывчатые вещества — галогениды азота (хлористый азот, иодистый азот).
С галогеноалканами аммиак вступает в реакцию нуклеофильного присоединения, образуя замещённый ион аммония (способ получения аминов):
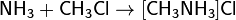 (гидрохлорид метиламмония)
(гидрохлорид метиламмония)
С карбоновыми кислотами, их ангидридами, галогенангидридами, эфирами и другими производными даёт амиды. С альдегидами и кетонами — основания Шиффа, которые возможно восстановить до соответствующих аминов (восстановительное аминирование).
При 1000 °C аммиак реагирует с углём, образуя синильную кислоту HCN и частично разлагаясь на азот и водород. Также он может реагировать с метаном, образуя ту же самую синильную кислоту:
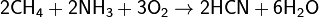
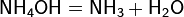
Получение. Промышленный способ получения аммиака основан на прямом взаимодействии водорода и азота:
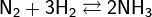 + 91,84 кДж
+ 91,84 кДж
Это так называемый процесс Габера (немецкий физик, разработал физико-химические основы метода).
Реакция происходит с выделением тепла и понижением объёма. Следовательно, исходя из принципа Ле-Шателье, реакцию следует проводить при возможно низких температурах и при высоких давлениях — тогда равновесие будет смещено вправо. Однако скорость реакции при низких температурах ничтожно мала, а при высоких увеличивается скорость обратной реакции. Проведение реакции при очень высоких давлениях требует создания специального, выдерживающего высокое давление оборудования, а значит и больших
капиталовложений. Кроме того, равновесие реакции даже при 700 °C устанавливается слишком медленно для практического её использования.
Применение катализатора (пористое железо с примесями Al2O3 и K2O) позволило ускорить достижение равновесного состояния. Интересно, что при поиске катализатора на эту роль пробовали более 20 тысяч различных веществ.
Учитывая все вышеприведённые факторы, процесс получения аммиака проводят при следующих условиях: температура 500 °C, давление 350 атмосфер, катализатор. Выход аммиака при таких условиях составляет около 30 %. В промышленных условиях использован принцип циркуляции — аммиак удаляют охлаждением, а непрореагировавшие азот и водород возвращают в колонну синтеза. Это оказывается более экономичным, чем достижение более высокого выхода реакции за счёт повышения давления.
Для получения аммиака в лаборатории используют действие сильных щелочей на соли аммония:
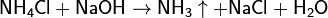
Обычно лабораторным способом аммиак получают слабым нагреванием смеси хлорида аммония с гашеной известью:
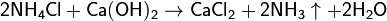
Для осушения аммиака его пропускают через смесь извести с едким натром.
Очень сухой аммиак можно получить, растворяя в нём металлический натрий и впоследствии перегоняя. Это лучше делать в системе, изготовленной из металла под вакуумом. Система должна выдерживать высокое давление (при комнатной температуре давление насыщенных паров аммиака около 10 атмосфер)[10]. В промышленности аммиак осушают в абсорбционных колоннах.
Выбор оптимальных условий проведения того или иного каталитического процесса в промышленных реакторах определяется различными факторами; решающее значение имеют, однако, термодинамические характеристики процесса.
Скорость реакции синтеза аммиака зависит от давления, температуры и состава реакционной смеси. Эта зависимость выражается уравнением Темкина-Пыжева с коэффициентом симметрии a - 0,5.
Концентрация аммиака. Скорость прямой реакции синтеза обратно пропорциональна парциальному давлению аммиака и, следовательно, его мольной доле, а для обратной реакции разложения характерна прямая пропорциональность. Таким образом, с повышением содержания аммиака общая скорость реакции падает (реакция “тормозится аммиаком”). Именно этим торможением объясняется повышение производительности процесса с ростом объемной скорости. Увеличение объемной скорости приводит к уменьшению прироста содержания аммиака и, тем самым, к увеличению средней скорости процесса. При низких температурах с повышением давления растет не только относительная степень превращения, но и абсолютное содержание аммиака в газовой смеси на выходе из реактора. Такой эффект влияния давления, возможно, тоже связан с торможением реакции водородом. По-видимому, при низких температурах значительная часть работающей поверхности заполнена хемосорбированным водородом. При высоких температурах выходы аммиака достигаются на смесях, близких к стехиометрическим, т.е. в условиях наибольших равновесных значений концентраций аммиака.
Давление. Скорость прямой реакции синтеза пропорциональна р1,5, а скорость обратной реакции - разложение аммиака - обратно пропорционально p0,5. С ростом давления наблюдаемая скорость реакции увеличивается. Как известно, образование аммиака из азота и водорода - реакция экзотермическая (DH500°C; 30 МПа = - 55,4 кДж/моль), сопровождается значительным уменьшением объема при постоянном давлении. Тем самым еще раз подтверждается вывод, что образованию аммиака благоприятствуют высокое давление и низкие температуры.
Инертные примеси. Как известно, производительность катализатора определяется, прежде всего, его химическим состоянием, макро- и микроструктурой, размером зерен, условиями восстановления и формирования. Зависит она от давления, температуры, объемной и линейной скоростей газового потока, состава газовой фазы по основным компонентам (азоту, водороду, аммиаку) и по примесям инертных газов (аргону, метану, гелию). Присутствие инертных примесей в смеси равносильно снижению общего давления, с увеличением их содержания скорость реакции уменьшается. Влияние инертных примесей на скорость реакции выражается (если отбросить второстепенные факторы) в том, что каждый процент этих примесей увеличивает эффективную мольную долю аммиака на 2%. А поскольку в первом приближении скорость реакции обратно пропорциональна эффективной мольной доле аммиака, то каждый процент инертных примесей уменьшает
скорость реакции на 2%.
Температура. Температура влияет на константы скоростей прямой и обратной реакции k1 и k2. Эту зависимость выражает закон Аррениуса.
Аммиакаты — продукты взаимодействия солей с аммиаком, комплексные соединения. Аммиакаты различаются как по составу [Ag(NH3)2]+, [Ni(NH3)4]2+, так и по устойчивости в водных растворах, используются в аналитической химии для обнаружения и разделения ионов металлов
Получение. Получают либо взаимодействием солей с NH3 в водном растворе, либо действием газообразного или жидкого NH3 на твердые соли. Например, аммиачный комплекс меди образуется в результате реакции:
Cu2+ + 4NH3 → [Cu(NH3)4]2+
Водный раствор аммиака при взаимодействии с ионами меди(II) сначала осаждает основные соли переменного состава зеленого цвета, легкорастворимые в избытке реагента. При этом образуется аммиачный комплекс меди сине-фиолетового цвета:
Cu2+ + 4NH4OH = [Cu(NH3)4]2+ + 4H2O
ГИДРАЗИН N2H4 (диамид), бесцв. дымящая на воздухе жидкость с неприятным запахом. Длина связи N—N 0,1449 нм, N—H 0,1021 нм; угол HNH 106°, NNH-1120, угол между плоскостями групп NH, 91° (гош-конформация); 6,04*10-30 Кл*м. Т. кип 113,5°С; давление пара 18,6 гПа (25°С); С° 98,83 Дж/(моль*К); 12,66 кДж/моль, 44,77 кДж/моль; So298 121,3 Дж/(моль*К); 0,90мПа*с; 66,7 мН/м; 0,625*105 Ом*м; 51,7; nD22 1,4695 (см. также табл.). Г. смешивается в любых соотношениях с водой, жидким NH3, спиртом; в неполярных р-рителях раств. плохо. С водой образует моногидрат, или гидразингидрат (т. пл. -51,6°С, т. кип. 118,5°С), тетрагидрат (т. пл. -80°С, т-ра эвтектики моногидрат-тетрагидрат -87°С), а также азеотроп (58,5 мол. % Г.; т. кип. 120,5°С). Жидкий Г. ассоциирован и слабо ионизован (2N2H4N2H5++N2H3-; К10-13). В нем хорошо раств. ми. соли, напр. LiCl, СаС12, NaNO3, NaClO4, Mg(ClO4)2. Большинство солей кристаллизуется из безводного Г. и гидразингидрата в виде прочных сольватов, напр. [Li(N2H4)2]ClO4, [Mg(N2H4)2](NO3)2. В таких комплексах молекулы Г. служат мостиковыми бидентатными или монодентатными лигандами. Энергия связи ионов непереходных металлов с Г. выше, чем с NH3 и водой. Г. термически малостабилен. Распад его в жидком и газообразном состояниях происходит с выделением тепла по ур-ниям:
При 200-300 °С в отсутствие катализаторов преобладает направление (1). Металлы ускоряют разложение паров Г., при этом их каталитич. активность изменяется в ряду Ir > Rh >. Родий, Pt и Pd направляют процесс по р-ции (2). В присут. остальных металлов ее вклад менее 10%. Г.-сильный восстановитель. Интенсивно окисляется О2 воздуха до N2, NH3, H2O; пар Г. горит синим пламенем. В щелочной среде окисляется медленно, ионы переходных металлов, особенно Сu2 +, ускоряют эту р-цию. Окисляется солями Се4+, Fe3+, Мn3+, Со3+ до NH3, N2 и воды, I2, Вr2, С12, ВrO3-, IO3-, IO4-, МnО4- до N2 и воды, Н2О2, HNO2, S2O82-, Mo(VI) до NH3, иногда до N2O. Нек-рые соли переходных металлов восстанавливаются Г. до металлов. В водном р-ре Г.-слабое основание, образующее одно-и двухзарядные ионы гидразония:
Известно большое число солей, отвечающих обеим ионным формам гидразония, св-ва Наиб. важных из них приведены в таблице. СВОЙСТВА ГИДРАЗИНА И СОЛЕЙ ГИДРАЗОНИЯ
* Плотн. твердого 1,146 г/см3. При алкилировании или арилировании Г. образуются алкил- и арилгидразины, при р-ции с карбонильными соед.— гидразоны и азины (см. Гидразина замещенные органические). Примеры неорг. производных Г., образующихся при замещении атомов Н,-гидразинсерная к-та N2H3SO3H, тетрафторгидразин N2F4. При действии амидов, гидридов или своб. металлов на безводный Г. образуются весьма взрывоопасные гидразиды, напр. NaHNNH2, используемые как реагенты в орг. синтезе. Будучи донором электронных пар, Г. может образовывать молекулярные комплексы с к-тами Льюиса (L) типа N2H4*L и N2H4*2L. Наиб. важные из них - кристаллич. гидразинборан N2H4*BH3 (—43 кДж/моль) и гидразинбисборан N2H4*2BH3 (- 127 кДж/моль). Оба соед., по зарубежным данным,-энергоемкое ракетное горючее. Г. получают окислением NH3 или CO(NH2)2 гипохлоритом Na:
Р-цию (1) проводят при 160°С и 2,5-3,0 МПа, р-цию (2)-при т-ре выше 100oС и нормальном давлении для предупреждения побочной р-ции: 2NH2C1 + N2H4 -> N2 + 2NH4C1 в смесь добавляют, напр., глицерин или желатину. Полученную смесь растворяют в 50%-ной H2SO4 и получают 2-3%-ные р-ры сульфата Г., к-рые затем обрабатывают NH3 и концентрируют (до содержания Г. 60%) дистилляцией при нормальном давлении или в вакууме; 70-90%-ный Г. получают, напр., экстрактивной дистилляцией с анилином, 1,2,6-гексатринитрилом, безводный Г.-обработкой гидразингидрата твердым NaOH с послед. перегонкой над NaOH, вымораживанием конц. водного р-ра Г., пиролизом цианурата N2H4*3HCNO при 147°С. Г. можно также получить р-цией элементного хлора с NH3 при — 30°С, илазмохимическим или радиохимическим разложением NH3. Обнаруживают Г. по образованию окрашенных соед. с нек-рыми альдегидами, напр. с диметиламинобензальдегидом. Г., 1,1-диметилгидразин и их смеси-горючие компоненты в ракетных топливах. Г. используют также как горючее в топливных элементах, ингибитор коррозии паровых котлов, для получения чистых металлов (Си, Ni и др.) из их оксидов и солей. Г., его соли и гидраты применяют: в произ-ве порообразователей (напр., бензолсульфонилгидразида), инсектицидов, ВВ, регуляторов роста растений (напр., гидразида маленновой к-ты), лек. ср-в (напр., противотуберкулезного ср-ва - гидразида изоникотиновой к-ты); как реактивы (в частности, для обнаружения карбонильных групп, пшохлоритов и хлоратов); для получения промежут. продуктов и красителей; в кач-ве добавок в стекломассу (напр., для устранения тусклости стекол); как реагенты для очистки пром. газов от СО2 и меркаптанов. Г. и его водные р-ры сильно ядовиты, раздражают слизистые оболочки, глаза и дыхат. пути, поражают центр. нервную систему и печень. При попадании Г. на кожу требуется немедленная ее обработка водой или слабым р-ром к-т; ПДК 0,1 мг/г3, пороговая концентрация, приводящая к миним. нарушениям высшей нервной деятельности, 0,02 мг/л, концентрация 0,0039 мг/л переносится 60 мин, 0,013 мг/л-10 мин, смертельная концентрация 1-2 мг/л. Пром. сточные воды, содержащие Г., обрабатывают С12 или хлорной известью либо пропускают через адсорберы с активным углем, стекловатой, золой, шлаком. Г. образует взрывоопасные смеси с воздухом и О2, способен к воспламенению в присут. асбеста, угля, оксидов Сu, Fe, Hg и др.; в воздухе т. всп. 270 °С (в чистом О2 и в присут. металлов и их оксидов т-ра вспышки понижается); нижний КПВ 4,7%
Гидроксиламин является важным промежуточным продуктом химической промышленности. Кроме того, в настоящее время концентрированные и высокочистые водные растворы гидроксиламина, почти не содержащие примесей (в частности, примесей металлов), находят применение в процессах очистки полупроводниковых плат.
Гидроксиламин и его соли являются токсичными веществами. Гидроксиламин при комнатной температуре обычно кристаллизуется. При нагревании кристаллы гидроксиламина становятся взрывоопасными, а водные его растворы очень нестабильны и быстро разлагаются. По этой причине обычно сначала получают растворы стабильных солей гидроксиламина, а растворы свободного гидроксиламина готовят непосредственно перед промышленным использованием, чтобы исключить их транспортировку. Однако при необходимости перевозки и/или хранения этих растворов возникает проблема подбора и введения стабилизирующих агентов. Разработаны стабилизаторы, предлагаемые для введения в растворы свободного гидроксиламина. В качестве таких стабилизаторов предлагается широкий спектр соединений, в частности [патент США 5783161, кл. С 01 В 021/20, опуб. 1998.07.21], аминопроизводные формулы N(А1R1)(А2R2)(А3R3), например, тиоацетамид, тиомочевина или 1,2-диаминоциклогексан-N,N,N’,N’-тетрауксусная кислота, и другие.

