 2015-04-20
2015-04-20 1658
1658Номенклатура показателей состава и загрязняющих свойств отходов бурения определяется в первую очередь областью их утилизации и требованиями к качеству природного объекта, обеспечивающего его нормальное функционирование как экологической системы [161]. Учитывая, что основными объектами природной среды, подверженными загрязнению отходами бурения, являются почвогрунты и водные системы, номенклатура показателей состава и загрязняющих свойств отходов бурения должна в полной мере отражать механизмы функционального повреждения экосистем таких природных объектов. В соответствии с этим определены показатели загрязняющих свойств отходов бурения [18,а]. Отрицательным воздействием на почвогрунты обладают следующие ингредиенты отходов бурения: нефть и нефтепродукты, трудноокисляемая органика, тяжелые металлы, минеральные соли. В составе последних необходимо индивидуально выделить ионы натрия, калия, хрома, гидрокарбонаты, хлориды и сульфаты как наиболее токсичные для почвогрунтов солевые компоненты. На рост растений и жизнедеятельность микроорганизмов непосредственно влияет показатель среды рН [139]. Резкое отклонение его значений от оптимального для почв различных типов может вызвать изменение соотношения органической и минеральной частей, направленности и скорости протекающих в почве химических, биологических и почвовосстанавливающих процессов [18, б].
Для водных экосистем, в частности для гидробионтов, наибольшую опасность, кроме нефти и нефтепродуктов, представляют взвешенные вещества (баритовый утяжелитель, известь, мелкие частички породы и др.) и коллоидные частицы минеральной и органической природы [49]. Они в основном проявляют токсический эффект, носящий механизм функциональных повреждений флоры и фауны. На физические параметры водных объектов значительное влияние оказывают полидисперсные взвеси различной природы, коллоидные вещества минеральной и органической природы, трудно- и легкоокисляемая органика, а также растворимые минеральные соли [141] и эвтрофицирующие агенты (фосфор, сера, азот) [190].
Эвтрофикация – обогащение рек и озер биогенными элементами, сопровождающееся повышением продуктивности вод.
В зависимости от направлений утилизации отходов номенклатура показателей их состава и свойств должна строго учитывать требования, предъявляемые к качеству таких материалов или их соединений. Так, например, в перечень показателей качества БСВ должны быть включены следующие дополнительные составляющие:
- химическое и биохимическое потребление кислорода, общая жесткость и щелочность, содержание аммонийного азота, фосфора, кальция, тяжелых металлов (при использовании сточных вод в ирригации или безопасном сбросе их на рельеф местности) [123];
- содержание сероводорода и железа (при использовании сточных вод в системе заводнения нефтяных пластов) [131].
При использовании ОБР и шлама в качестве вторичных сырьевых ресурсов в производстве изделий грубой строительной керамики основными являются показатели элементного состава [185], т.е. для каждого направления утилизации отходов бурения номенклатура показателей ограничивается требованиями не только загрязняющих свойств (главным образом, санитарно-токсикологической направленности), но и потребительских свойств готовой промышленной продукции.
Номенклатура основных показателей состава и загрязняющих свойств отходов бурения и необходимость их определения применительно к различным природным объектам приведены в табл. 3.11.
Таблица 3.11
Номенклатура показателей состава и свойств отходов бурения
| Показатель | БСВ | ОБР | БШ | ||
| системы оборотного водоснабжения | Системы ППД | Системы сброса на рельеф местности | |||
| Плотность | - | - | - | + | + |
| Активность ионов водорода (рН) | + | + | + | + | + |
| Химическое потребление кислорода (ХПК) | + | - | + | + | |
| Биохимическое потребление кислорода (БПК) | - | - | + | - | - |
| Растворенный кислород (РК) | - | + | - | - | - |
| Фазовый состав | |||||
| В том числе: | |||||
| твердой фазы | - | - | - | + | + |
| жидкой фазы | - | - | - | + | + |
| нефти и нефтепродуктов | - | - | - | + | + |
| Содержание нефтепродуктов (НП) | + | + | + | + | + |
| Содержание растворенных органических и неорганических веществ (сухой остаток — СО) | + | + | + | - | - |
| Содержание растворенных неорганических веществ ПО) | + | + | + | - | - |
| Содержание взвешенных веществ (ВВ) | + | + | + | - | - |
| Жесткость (Ж) | + | + | + | + | + |
| Щелочность (Щ) | + | + | + | + | + |
| Содержание ионов: | |||||
| Ca2+ | + | + | + | + | + |
| Mg2+ | + | + | + | + | + |
| Feобщ | + | + | + | + | + |
| Sn2 + | - | - | + | + | + |
| Pb2 + | - | - | + | + | + |
| Cm3+ | - | - | + | + | + |
| Ва2+ | - | - | + | + | + |
| Na+, К+ | - | - | + | + | + |
| Cl- | + | + | + | + | + |
| SO42- | + | + | + | + | + |
| Содержание азота аммиачного | - | - | + | - | - |
| Содержание Р2О5 | - | - | + | - | - |
| Содержание H2S | - | + | - | - | - |
Методы и техника анализа загрязнителей органической природы
Органические вещества в промышленных стоках можно определить либо неспецифическими методами (общее содержание органических компонентов), либо методами группового анализа (классификация веществ по наличию определенных функциональных групп), либо методами идентификации отдельных органических веществ, выделенных из общей смеси и сконцентрированных до необходимого содержания интересующего компонента [165]. Определение сотен индивидуальных органических веществ в короткое время нереально, в связи с чем задерживаются информация о загрязнении и меры по предотвращению загрязнений [18, а, 171, а].
Поэтому необходимыми величинами при определении общесанитарного показателя вредности, по которым нормируется примерно 15 % от общего количества нормируемых веществ [164], являются такие общие показатели качества вод, как биохимическое и химическое потребление кислорода и содержание общего органического углерода.
Химическое потребление кислорода (ХПК).В зависимости от степени загрязнения жидкие отходы бурения содержат значительные количества легкоокисляющихся веществ: органических и некоторых неорганических. Количество кислорода, эквивалентное расходу окислителя, характеризует значение окисляемости. Если устранить влияние мешающих неорганических примесей (закисного железа, нитратов, сероводорода), то результаты определения окисляемости дают косвенное представление о содержании в пробе органических веществ. В зависимости от применяемого окислителя различают окисляемости перманганатную, бихроматную и др.
Перманганатный метод, ранее широко применявшийся, совершенно не пригоден для анализа сильно загрязненных стоков, к которым относятся буровые растворы и буровые сточные воды: перманганат калия - недостаточно сильный окислитель, и органические вещества окисляются не полностью.
Унифицированным является определение ХПК с использованием бихромата калия. Этот окислитель совместно с катализатором - сернокислым серебром - окисляет практически все органические соединения. Получаемое при этом значение ХПК составляет 95 — 98 % от ХПК теоретического [99]. Сущность метода сводится к окислению органических веществ раствором бихромата калия в присутствии концентрированной серной кислоты и катализатора - сернокислого серебра. Мешающее влияние хлорид-ионов устраняется введением в пробу сульфата двухвалентной ртути (II). После окисления избыток бихромата определяют титрованием раствором соли Мора. Метод трудоемкий и длительный. Только кипячение пробы с окислителем длится 2 ч.
В практику ежедневных анализов по определению органических веществ широко вошли ускоренные методы определения ХПК, которые сводятся к уменьшению времени нагревания или повышению концентрации серной кислоты, за счет чего нагревания извне не требуется. Результаты определения получаются несколько заниженными, но хорошо воспроизводимыми. Периодическое определение обоими методами (ускоренным и арбитражным) позволяет ввести приблизительный коэффициент пересчета.
Анализ развивающихся инструментальных физико-химических методов определения ХПК [165] приводит к тому, что основная направленность физико-химических методов сводится к определению непрореагировавшего бихромата калия с помощью ИК-спектрометров, фотометрических и электрохимических анализаторов. Высокая чувствительность этих методов, безусловно, позволит внедрить их в практику определения ХПК при условии сокращения продолжительности определений. Чаще всего объектами контроля при исследовании буровых стоков являются мутные и окрашенные воды и фильтраты. Это накладывает ограничение на применение фотометрического метода определения вышеуказанного показателя. Применяемым является потенциометрический контроль над расходом окислителя, который в принципе дублирует унифицированную методику и дает хорошую корреляцию данных с теоретически рассчитанным значением окисляемости [99].
Биохимическое потребление кислорода (БПК).Под биохимическим потреблением кислорода понимается количество кислорода (в миллиграммах), необходимое для окисления органических веществ, находящихся в 1 л сточной воды, при биологических процессах в аэробных условиях. В значение БПК не входит расход кислорода на нитрификацию, протекающую под действием разных бактерий. Биохимическое окисление различных органических веществ происходит с разной скоростью. К легкоокисляемым, биологически мягким веществам относятся фенол, фурфурол, анионоактивные поверхностно-активные вещества и др. [99]. К медленно разрушающимся, биологически жестким веществам относятся сульфанол НП-1, неионогенные ПАВ и др. Так как стоки буровых предприятий содержат в своем составе в основном неионогенные ПАВ типов ОП-10, ОП-4, ОП-7 и сульфонол НП-1, допускается определение только БПК полного.
Сущность метода заключается в следующем. Исследуемую пробу разбавляют чистой водой; определив содержание растворенного кислорода, ее оставляют в склянке на 2, 3, …, 10 сут. Через эти промежутки времени содержание кислорода определяют вновь. Когда поглощение кислорода в пробе прекратится, то количество кислорода, использованное на окисление и отнесенное к 1 л пробы, будет значением БПК. Для подавления нитрификации в пробу вводят специальные вещества, подавляющие жизнедеятельность нитрифицирующих микроорганизмов и при этом абсолютно не влияющие на микроорганизмы, которые осуществляют основной биохимический процесс окисления.
Следить за ходом биохимического окисления можно не только по уменьшению количества кислорода, но и по уменьшению содержания органических веществ (по показателю ХПК). Соответственно существует два метода определения БПК: метод разбавления и метод определения по разности между результатами определения ХПК. Предварительная подготовка микрофлоры и обработка проб по обоим методам аналогичны. Поэтому различие методов существует лишь в определении окончательного параметра ХПК или растворенного кислорода. В литературе не имеется данных о корреляции этих параметров. В связи с тем, что определение ХПК для отходов бурения является обязательным, по-видимому, целесообразно и определение БПК проводить по второму методу, не прибегая к отработке довольно сложной методики определения растворенного кислорода.
В том случае, когда по нормативным требованиям для утилизации очищенных БСВ, например для заводнения нефтяных пластов, требуется определение растворенного кислорода, его следует проводить универсальным иодометрическим методом Винклера [89, 99]. В основе метода лежит реакция окисления марганца (II) в щелочной среде растворенным кислородом. Образующийся осадок гидроксида марганца (IV) бурого цвета при последующем подкислении в присутствии иодида калия растворяется с образованием свободного йода, который определяют титрованием тиосульфатом натрия. Для нефтепромысловых вод, характеризующихся высокой минерализацией, наличием растворенного сероводорода, нефтепродуктов и химических реагентов, применяемых для интенсификации нефтедобычи разработаны новые условия хода анализа [92]. Минимально определяемая концентрация растворенного кислорода составляет 0,1 мг/л. Погрешность анализа в диапазоне концентраций кислорода 0,1- 0,4 мг/л не превышает ±20%; 0,4 - 1,0 мг/л - ± 10 %; больше 1 мг/л - ± 5 %. Продолжительность анализа составляет 2,5 ч.
Общий органический углерод (ООУ). Часто результаты косвенного определения общего содержания органических веществ по окисляемости, полученные различными методами, несопоставимы. Поэтому определение окисляемости рекомендуют заменять или дополнять определением ООУ. Считают метод определения ООУ наиболее перспективным, вытесняющим устаревшие способы оценки загрязняющих свойств отходов бурения [56].
Определение ООУ основано на аналитических методах: жидкофазном окислении с помощью перманганата, персульфата или бихромата калия; каталитическом окислении в токе воздуха или кислорода; сжигании при температуре 900 — 1200 °С без катализатора. Наиболее часто применяемые методы определения ООУ по принципу деструкции органических веществ можно разделить на три основные группы: сухое термическое разложение органических веществ с последующим окислением продуктов пиролиза до диоксида углерода, мокрое низкотемпературное окисление с применением сильных окислителей и фотохимическое разложение органических веществ под действием жесткого УФ-излучения. Результаты определения выражают не в количестве кислорода, необходимого для окисления органических веществ, а непосредственно в содержании углерода. Окисление обычно идет до выделения С02 или СН4, которые определяются ИК-спектрометрами и другими современными анализаторами [173]. Автоматический анализ для определения малых количеств органических соединений делает этот метод перспективным. Однако уровень оснащенности аналитических лабораторий отрасли, постоянно изменяющийся состав стоков буровых предприятий, ненормируемость показателя ООУ для различных направлений утилизации отходов бурения не позволяют применять ООУ для оценки содержания органических веществ в отходах бурения. На данном этапе предпочтение следует отдать показателю ХПК, тем более что значения ХПК, полученные бихроматным методом, хорошо коррелируют с данными определения ООУ.
Нефть и нефтепродукты (НП). В понятие "нефтепродукты" входят неполярные и малополярные соединения, экстрагируемые н-гексаном (или петролейным эфиром) [173]. Это ограничивает понятие "нефтепродукты" углеводородами (алифатическими, алициклическими, ароматическими), являющимися основной составной частью нефти. Нафтеновые кислоты и фенолы в результате определения не входят в это понятие и устанавливаются отдельно.
Для определения летучих нефтепродуктов (бензиновые и частично керосиновые фракции) принят метод, основанный на отгонке, последующей конденсации и гравиметрическом окончании с использованием перегонного аппарата со специальной калиброванной ловушкой [173]. Анализ проводится при общем содержании нефтепродуктов в пробе не менее 10 мг/л. Метод утвержден в качестве арбитражного и обладает чувствительностью 5 мг/л. Основное его достоинство в том, что исключается приготовление стандартных растворов такого же качественного и количественного состава, как и анализируемый раствор.
Наиболее распространенным, хотя и трудоемким методом определения НП, является весовой метод, признанный арбитражным. Сущность его сводится к экстракции нефтепродуктов из стоков хлороформом или четыреххлористым углеродом, отгонке растворителя, последующему растворению остатка в гексане, удалению полярных соединений и гравиметрическому определению растворенных в гексане веществ. Диапазон измерения НП составляет от 0,3 до 3 мг/л. При большем содержании нефтепродуктов уменьшают объем пробы. Для анализируемых проб, содержащих менее 0,3 мг/л нефтепродуктов, следует ввести определение с использованием газожидкостной хроматографии (ГЖХ), основанной на экстракции нефтепродуктов из сточных вод растворителем (н-гексаном, пентаном, четыреххлористым углеродом) и последующем газохроматографическом исследовании [198]. Этим методом можно определять суммарное содержание, а также типы нефтепродуктов в сточных водах. Чувствительность при применении пламенно-ионизационного детектора — 0,1 мг/л всех углеводородов, 0,005 мг/л каждого углеводорода. Относительная ошибка определения 3-10%. Селективность нефтепродуктов в пробе производится по температурам кипения углеводородов, а также числу атомов в молекуле. В зависимости от используемого растворителя возможно определение углеводородов с температурой кипения от 85 до 450 °С. На современных хроматографах хроматограмма записывается в виде сигнала детектора, который пропорционален концентрации определяемого компонента от времени. Квалифицированный работник, пользуясь известными данными о температурах кипения нормальных парафиновых углеводородов и идентифицируя пики хроматограмм по отвечающим им числам атомов углерода в молекуле, судит о том, каким веществом (сырой нефтью, дизельным топливом и т.п.) загрязнена сточная вода.
Если непосредственно невозможно идентифицировать пики нормальных парафинов (вследствие их очень низкого содержания или из-за отсутствия опыта у исследователя), то готовят синтетическую смесь известных н-парафинов примерно с теми же точками кипения, как у парафинов пробы. Затем вводят раствор этой смеси в определенном соотношении в пробу и хроматографируют в тех же условиях, что и в первый раз. Сравнением обеих хроматограмм точно устанавливают принадлежность тех или иных пиков к нормальным парафинам.
Для количественного определения содержания любого нефтепродукта используют метод градуировочного графика, т.е. получают хроматограмму любого нефтепродукта в используемом растворителе точно известной концентрации. Этот раствор не должен содержать других веществ, помимо углеводородов, с температурами кипения приблизительно в тех же границах, что и углеводородов анализируемой пробы. Сравнением площадей пиков определяемого компонента и сравнительного нефтепродукта находят количественное содержание нефтепродукта в пробе. (Детальное описание арбитражного ГЖХ-метода приведено для того, чтобы показать необходимость работы на хроматографе высококвалифицированных специалистов. Тогда выявятся все достоинства этого метода: высокая чувствительность, скорость и точность определений любого ингредиента нефтепродуктов).
Наиболее эффективно применение этого метода для анализа технологических процессов, сопровождающихся образованием нефтесодержащих стоков с приблизительно одинаковым качественным составом нефтепродуктов. Применение этого метода для единичных анализов стоков различных месторождений менее целесообразно.
Нефтепродукты отделяют от других веществ, извлекаемых хлороформом, в тонком слое силикагеля при использовании гексана в качестве подвижного растворителя. Хроматографическое отделение и концентрирование нефтепродуктов проводят в тонком слое. Определение заканчивают турбидиметрическим методом, т.е. измерение оптической плотности эмульсии нефтепродуктов в водно-желатиновом растворе. Просмотр хроматограммы в УФ-свете дает возможность приближенно оценить содержание нефтепродуктов в анализируемой пробе, а также полноту их отделения от других органических веществ, извлеченных хлороформом. Чувствительность метода - 0,3 мг во взятом для анализа объеме пробы. Погрешность определения - ± 10 %. Достоинство метода в том, что в связи с малой зависимостью результатов турбидиметрических определений от углеводородного состава нефтепродуктов не требуется подготовки стандартных растворов для каждого вида анализируемых проб. Недостатки метода - трудоемкость и продолжительность определения.
Снизить время проведения анализа одной пробы до 15 - 20 мин. позволяет ускоренный адсорбционно-люминесцентный метод, основанный на выделении НП из воды четыреххлористым углеводородом, отделении полярных соединений сорбцией на окиси алюминия и измерении люминесценции раствора нефтепродуктов на флуориметре при определенной длине волны. Содержание НП в пробе находят по показанию шкалы прибора, прокалиброванного по стандартному раствору. Недостаток метода в том, что стандартный раствор должен быть приготовлен из нефтепродуктов этого же источника загрязнения, т.е. для каждого вида анализируемых проб необходимо готовить свой стандартный раствор. Метод к тому же не является арбитражным, т.е. необходима периодическая сверка получаемых результатов с результатами определения арбитражными методами и введение соответствующей поправки.
В отличие от ранее рассмотренного арбитражный метод ИК-спектрометрии не требует приготовления специального стандартного раствора для каждого вида вод. При построении калибровочного графика используют смесь, состав которой соответствует составу наиболее распространенных нефтей нашей страны. В ИК-спектрах имеются соответствующие отклонения вследствие различий в содержании метильных и метиленовых групп в разных по составу нефтепродуктах. Однако эти отклонения невелики и при использовании указанной смеси для построения калибровочного графика ошибки определения незначительны. Возможны два варианта такой смеси: а) если сточная вода заведомо не содержит легколетучих нефтепродуктов, смесь готовят в процентах к объему: гексана - 59, изооктана - 2, бензола - 39; б) если проба содержит и легколетучие углеводороды, смесь составляют в процентах к объему: гексана - 51, изооктана - 5 и бензола - 44.
Поскольку составы этих смесей постоянны и зависимость между оптической плотностью и концентрацией нефтепродуктов линейна, можно избежать построения калибровочного графика при выполнении каждого анализа. Достаточно воспользоваться экспериментально найденными значениями этого коэффициента: К1 = 0,437 (для вод, содержащих летучие нефтепродукты) и К2 = 0,542 (для стоков, не содержащих летучих нефтепродуктов), т.е. значительно упрощается не только окончание определения, производимого на спектрометре, но и вычисление показателя НП [99].
Начальный ход анализа аналогичен ранее описанным методам: экстракция нефтепродуктов четыреххлористым углеродом, причем без предварительного удаления летучих составляющих, отделение полярных соединений на окиси алюминия и далее снятие ИК-спектра полученного раствора с измерением оптической плотности при волновом числе 2926 см -1. Лабораторные исследования физико-химических свойств БСВ подтверждают наибольшую надежность и объективность весового и особенно ИК-спектрометрического метода определения нефтепродуктов.
Для быстроты предварительного анализа в ИК-спектрометрическом методе используют люминесцентный метод. Созданный на основе фотолюминесцентного метода флуориметр "Квант" прост в обслуживании, с его применением разработаны и внедрены инструментальные методики определения нефтепродуктов в промышленных сточных водах.
При контроле технологических показателей бурового раствора в процессе бурения скважин содержание нефти определяют либо методом экстракции (прибор Дина-Старка), либо выпариванием жидкой фазы из заданного объема бурового раствора (установка ТФН-1). В определяемые этими методами органические соединения входят полярные соединения и часть ароматических, так как в качестве растворителя используют ксилол. Это противоречит самому понятию "нефтепродукты", приведенному выше. Поэтому эти методики целесообразно использовать при исследовании фазового состава ОБР и БШ, а не для определения показателя НП.
Методы и техника анализа загрязнителей неорганической природы. Хлориды. Для анализа стоков с небольшим содержанием хлоридионов (1,0 - 1,5 мг/л) служит нефелометрический метод определения, основанный на образовании хлорида серебра и последующем визуальном сравнении мутности пробы с мутностью эталонов стандартной шкалы. Если концентрация хлоридов меньше 3 мг/л, более точным и чувствительным оказывается фотометрический метод с применением дифенилкарбазида [173]. Сущность метода сводится к образованию хлорид-ионами с ионами ртути (II) малодиссоциированного хлорида ртути. В присутствии дифенилкарбазида образуется окрашенное в фиолетовый цвет комплексное соединение, интенсивность окраски которого пропорциональна количеству хлоридов. Спектрофото- или фотоколориметрическое окончание определения по заранее приготовленной калибровочной кривой предпочтительнее визуального. Однако для определения хлоридов в стоках бурения, содержащих иногда более 13,0 мг/л ионов хлора [173], наиболее применимы меркури-, аргентометрический по Мору и ионометрический методы.
Меркуриметрическое определение сводится к образованию чрезвычайно малодиссоциированного хлорида ртути (II). Индикатор дифенилкарбазон образует с избытком ионов ртути соединение фиолетового цвета. Определение проводят титрованием в азотнокислой среде при рН = 2,5 ± 0,1. Резкость изменения окраски индикатора в значительной мере зависит от соблюдения правильного значения рН. При рН = 2,0 индикатор не окрашивается, а при рН > 3,0 появление окраски запаздывает. Это значительный недостаток метода, так как буровые стоки в основном имеют нейтральную и щелочную реакции. Без разбавления и упаривания этим методом определяют хлориды концентрацией 10 - 1000 мг/л.
Диапазон определяемой концентрации хлоридов аргентометрическим методом более узкий (от 2 до 400 мг/л). Сущность метода сводится к осаждению растворенного хлорид-иона азотнокислым серебром в виде малорастворимого хлорида серебра. Хромат калия, применяемый в методе Мора в качестве индикатора, реагирует с избыточными ионами серебра с изменением окраски от лимонно-желтой до оранжево-желтой при рН = 7 ÷ 10. По Фольгарду, к анализируемому раствору добавляют избыток азотнокислого серебра, осадок хлорида серебра отфильтровывают и определяют избыток нитрата серебра титрованием роданида калия в присутствии железоаммонийных квасцов в качестве индикатора. По всем указанным методам одновременно с хлорид-ионами титруются бромид-, иодид-, цианид- и роданид-ионы. Если геолого-технические условия проводки скважин и применяемые для обработки буровых растворов реагенты, не способствуют внесению этих компонентов в отходы бурения, то содержанием этих примесей можно пренебречь и воспользоваться аргентометрическим или меркуриметрическим определением. Окрашенные органическими соединениями сильно загрязненные сточные воды перед определением выпаривают в щелочной среде досуха и слегка прокаливают. Мешающие определению взвешенные вещества удаляют фильтрованием пробы, а небольшие количества сульфит-, тиосульфит-, сульфид-, роданид- и цианид-ионов окисляют пероксидом водорода в щелочной среде. В меркуриметрическом методе мешающими являются также ионы железа (III), цинка, свинца, алюминия, никеля и хрома (III). Это следует учитывать при анализе очищенных методом реагентной коагуляции сточных вод, так как в качестве коагулянтов часто применяют алюминий- и железосодержащие реагенты.
При наличии в лабораториях ионоселективных электродов возможно применение метода прямой ионометрии для определения фторидов, иодидов и хлоридов в пробах. Сущность метода (ГОСТ 26425 - 85) заключается в определении разности потенциалов хлоридного ион-селективного и вспомогательного электродов, значение которой зависит от концентрации иона хлорида в растворе. В качестве вспомогательного используют насыщенный хлорсеребряный электрод. Метод хорошо отработан для определения иона хлорида в водной вытяжке почвы и может с успехом применяться для анализа водной вытяжки шлама и сильно загрязненного органическими веществами бурового раствора.
Ионометрически с использованием хлоридного электрода можно проводить и титрование пробы раствором азотнокислого серебра. Для этого применяют рН-метры с блоком автоматического титрования с погрешностью измерений не более 5 мВ.
Определение иона хлора этим методом отличается удобством и быстротой. Допускаемые относительные отклонения при выборочном статистическом контроле составляют: для количества эквивалентов ионов хлорида до 2 ммоль в 100 г сухого образца - 21 %; свыше 2 до 6 ммоль - 11 %; свыше 6 ммоль - 7 %. Для определения ионов Br -, I-, F-, Сl- разработан ряд новых методов (хемилюминесцентный, флуоресцентный, электрохимический), которые перспективны для анализа вод благодаря высокой чувствительности и селективности. Однако в отечественной практике анализа сточных вод они пока не нашли широкого распространения.
Сульфаты. Необходимость разработки надежных и простых методов определения сульфатов обусловлена тем, что они являются важнейшим компонентом большинства природных и техногенных объектов. Методы определения сульфатов в водных средах сводятся к осаждению иона раствором хлористого бария в кислой среде в виде малорастворимого сернокислого бария. В зависимости от способа количественного определения полученного осадка существуют следующие методы.
При тетриметрическом определении сульфатов по ГОСТ 26426 - 85 осадок сульфата бария отфильтровывают, промывают, растворяют в щелочном растворе трилона Б и титруют избыток последнего хлоридом магния. Метод применяется при содержании сульфат-ионов от 5 до 25 мг/л. Гравиметрическое определение основано на взвешивании отмытого и прокаленного при температуре не выше 800 °С осадка сульфата бария. Оба метода трудоемки, длительны. Определению мешают взвешенные вещества, кремниевая кислота, ионы железа (III) и бихроматы. В комплексонометрическом методе к перечисленным, мешающим определению веществам добавляются еще все катионы, реагирующие с трилоном Б. Поэтому необходим комплекс мероприятий (фильтрование, прибавление активированного угля, пропускание раствора через колонку с катионитом и др.) для устранения влияния примесей.
Используют также метод прямого титрования нитратом свинца в присутствии дитизона. Прямое титрование можно осуществлять и раствором азотнокислого бария. Метод чувствительный и точный, однако мешающее влияние могут оказывать фосфат-ионы, хроматы, иодины и другие анионы.
Фотоколометрический метод определения сульфатов позволяет определять от 2 до 2000 мг/л сульфатов в природных и сточных водах, а также почвенных вытяжках. Определению мешают взвешенные вещества и мутность, которые удаляются фильтрованием.
Среди инструментальных методов определения сульфатов перспективна потенциометрия с использованием ион-селективных электродов. Особенно ценятся такие свойства электродов, как быстродействие, возможность работы в широком диапазоне концентраций и в мутных окрашенных растворах. С точки зрения аппаратурного оформления установки для проведения потен-циометрического титрования пробы могут быть легко автоматизированы.
До настоящего времени отечественные промышленные образцы ион-селективных электродов с сульфатной функцией отсутствуют. Поэтому следует использовать Pb-селективный электрод с твердой мембраной из сульфида свинца и сульфида серебра [188]. Установлено, что в этом методе мешающего влияния не оказывают ионы кальция, магния, натрия, хлора при следующих соотношениях: S 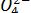 : Са2 + = 1: 5; S : Mg2 + = 1: 4; S : Na+ = 1: 10; S : Сl- = 1:10. Влияние карбонат-иона установлено при рН = 6, поэтому определение сульфата следует вести в интервале рН 2,5 - 4,5. Метод дает хорошие результаты при определении сульфатов в минеральных водах. На одно титрование затрачивается 2 - 3 мин. и не требуется предварительной подготовки анализируемой пробы. Относительная ошибка определения не превышает 1 %.
: Са2 + = 1: 5; S : Mg2 + = 1: 4; S : Na+ = 1: 10; S : Сl- = 1:10. Влияние карбонат-иона установлено при рН = 6, поэтому определение сульфата следует вести в интервале рН 2,5 - 4,5. Метод дает хорошие результаты при определении сульфатов в минеральных водах. На одно титрование затрачивается 2 - 3 мин. и не требуется предварительной подготовки анализируемой пробы. Относительная ошибка определения не превышает 1 %.
Для определения сульфатов целесообразно использовать барий селективные электроды с пластифицированной отвержденной мембраной [79]. Разработаны две методики определения: метод добавок и метод дифференциального титрования. Обе дают хорошую воспроизводимость для природных и питьевых вод. Перечисленные преимущества потенциометрического титрования делают его перспективным для определения сульфатов в буровых сточных водах, хотя отработанной методики пока не существует.
Щелочность. Этот показатель - один из наиболее важных для определения загрязняющих свойств отходов бурения. Щелочностью называют содержание в водных растворах веществ, вступающих в реакцию с сильными кислотами, т.е. с ионами водорода. К таким веществам относят сильные основания, диссоциирующие в разбавленных растворах с образованием гидроксид-ионов (NaOH, КОН и т.д.); слабые основания (аммиак, пиридин и т.д.) и анионы слабых кислот(НС 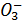 , С
, С 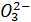 , Si , Н2Р
, Si , Н2Р 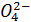 , HS , S
, HS , S 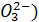 ; анионы гуминовых кислот (HS-, S2- и т.д.). Веществ, титруемых сильными кислотами, много, поэтому показатель щелочности недостаточно характеризует ее компонентный состав. При определении этого показателя необходимо знать предварительный качественный состав стоков: реагенты, которыми обрабатывали буровой раствор в процессе бурения, или буровую сточную воду при ее очистке; перечень веществ, попадание которых в местах сбора стоков было возможным, состав тампонажных и обезвреживающих смесей. В связи с тем, что отходы бурения содержат спектр ионов, влияющих на показатель щелочности, единицей измерения его следует считать мг-экв/л. Пересчет щелочности, выраженный в мг-экв/л, на содержание ионов карбонатов, бикарбонатов и гидроксилов, как это принято в анализе почв по ГОСТ 26424 - 85 и природных вод, приводит к искаженным количественным результатам.
; анионы гуминовых кислот (HS-, S2- и т.д.). Веществ, титруемых сильными кислотами, много, поэтому показатель щелочности недостаточно характеризует ее компонентный состав. При определении этого показателя необходимо знать предварительный качественный состав стоков: реагенты, которыми обрабатывали буровой раствор в процессе бурения, или буровую сточную воду при ее очистке; перечень веществ, попадание которых в местах сбора стоков было возможным, состав тампонажных и обезвреживающих смесей. В связи с тем, что отходы бурения содержат спектр ионов, влияющих на показатель щелочности, единицей измерения его следует считать мг-экв/л. Пересчет щелочности, выраженный в мг-экв/л, на содержание ионов карбонатов, бикарбонатов и гидроксилов, как это принято в анализе почв по ГОСТ 26424 - 85 и природных вод, приводит к искаженным количественным результатам.
В практике анализа состава БСВ принята единственная методика определения щелочности - титрование пробы сильной кислотой в присутствии двух индикаторов, интервалы изменения окраски которых находятся в разных областях рН. Потенциометрическое титрование удобно вести с использованием блока автоматического титрования, например, типа БАТ-15. При потенциометрическом титровании (т.е. при непрерывном измерении рН раствора) на кривой "объем прибавленной кислоты - рН раствора" получится несколько точек эквивалентности. Они соответствуют содержанию различных классов веществ, обусловливающих щелочность, что способствует определению конкретных компонентов сточных вод. Кроме того, потенциометрическое титрование незаменимо для анализа окрашенных проб.
Натрий и калий. Пламефотометрический метод определения ионов натрия и калия является разновидностью эмиссионного спектрального анализа [173] и основан на изменении интенсивности излучения атомов (реже молекул), возбужденных в пламени. Вследствие возбуждения изменяется окраска пламени солями натрия и калия.
Количественный анализ основан на прямой зависимости сигнала эмиссии от концентрации определяемого элемента. Для проведения анализа применяют пламенный фотометр с монохроматором или интерференционными светофильтрами с максимумом пропускания в области 588 - 590 нм для определения натрия и 766 - 770 нм для калия. Чувствительность определения колеблется от 0,001 до 5,0 мг/л и зависит от чувствительности используемой марки пламенного фотометра. Суммарная относительная погрешность метода составляет 7,5 % при определении натрия и 10 % при определении калия.
Более трудоемкими, длительными по времени являются следующие методы определения натрия.
Гравиметрический метод основан на осаждении натрия в виде натрийцинкуранилацетата. Мешающее влияние оказывают литий, фосфаты, некоторые органические кислоты.
Титриметрический метод основан на комплексонометрическом определении уранила, используемого для осаждения ионов натрия. Эти анализы многоступенчатые и длительные, используется труднодоступный реагент.
Широкое применение в санитарно-химическом анализе объектов окружающей среды находит полярографический метод. Полярографированию поддаются практически все катионы металлов, многие анионы, неорганические и органические вещества, способные к электрохимическому окислению или восстановлению. Определение щелочных и щелочно-земельных элементов осуществляется на фоне тетраал-киламмониевых соединений, адсорбирующихся на поверхности ртутного электрода, со значительным повышением потенциала выделения водорода. Определению ионов натрия мешают ионы кальция, магния и железа, однако под воздействием гидроокиси тетраметиламмония, служащего фоном, происходит осаждение этих ионов. Ион калия восстанавливается при потенциале, близком к потенциалу восстановления натрия. При соотношении К+: Na+ = 8 калий не мешает определению. При больших количествах калия его осаждают дипикриламинатом магния. Полярографический метод определения различных ионов обладает рядом преимуществ: нижний предел определяемых концентраций составляет 10-5 - 10-6 кмоль/л; возможно установление качественного и количественного состава пробы без предварительного разделения ряда веществ.
Колориметрический метод определения калия основан на окислении дихроматом выделенного в осадок гексанитрокобальта (III) натрия и калия с последующим определением интенсивности окраски раствора на фотоэлектроколоримет-ре или визуально в цилиндрах Несслера. Обязательным условием проведения анализа являются фильтрование пробы и концентрирование ее при содержании калия менее 100 мг/л. Анализу мешают ионы аммония, кремнекислота и органические вещества.
Титриметрический метод определения калия основан на растворении осадка тетрафенилбората калия в ацетоне с последующим аргентометрическим его определением в исследуемой пробе. Тетрафенилборат серебра практически нерастворим в ацетоне. Мешающее влияние оказывают ионы аммония, которые можно удалить кипячением после добавления щелочи натрия или предварительным прокаливанием сухого остатка. Существенным недостатком методов являются длительность, многостадийность.
Анализируя предложенные методы определения калия, следует сделать заключение: наилучший во всех отношениях - метод пламенно-эмиссионной спектрометрии.
Кальций. Соли кальция — постоянная составная часть поверхностных, грунтовых и сточных вод различных производств. Определение кальция методом атомно-абсорбционной спектрометрии (ААС) основано на поглощении УФ или видимого излучения атомами газа. Чтобы перевести пробу (хотя бы частично) в газообразное атомное состояние, ее впрыскивают в пламя. В качестве источника излучения применяют лампу с полым катодом из определенного металла. Интервал длин волн спектральной линии, испускаемой источниками света, и линии поглощения того же самого элемента в пламени очень узок, поэтому мешающее поглощение других элементов практически не сказывается на результатах анализа.
В принципе ААС подобна обычной спектрофотометрии, аналогична и используемая в обоих методах аппаратура. Излучение пропускают через анализируемую пробу, которая частично его поглощает, а пропущенный свет проходит через монохроматор и попадает на фотодетекторрегистрирующее устройство, отмечающее количество пропущенного или поглощенного света. Различия этих методов — в источнике света и в кювете для пробы.
Источником света в ААС служит лампа с полым катодом, испускающая свет, имеющий очень узкий интервал длин волн порядка 0,001 нм. Прибор содержит необходимый набор ламп, каждая лампа предназначена для определения только одного какого-либо элемента. Кюветом в ААС служит само пламя, для получения которого обычно, если нет специальных указаний, используется воздушно-ацетиленовая газовая смесь.
При анализе сточных вод, как правило, требуется разбавление пробы.
Приборы для атомно-абсорбционной спектрофотометрии различаются и по конструкции, и по методике работы на них, поэтому следует строго следовать прилагаемой к прибору инструкции.
Прямое определение кальция возможно, когда его концентрация превышает 100 мкг/л.
При определении содержания кальция в шламе или в почве суммарная относительная погрешность для ААС, выраженная коэффициентом вариации, составляет, %: для количества эквивалентов кальция от 0,5 до 2 ммоль в 100 г почвы - 12,5; от 2 до 6 ммоль - 10; свыше 6 ммоль - 6 (ГОСТ 26428 - 85).
Наиболее распространен комплексонометрический метод определения кальция в пробе, в которой присутствуют соли магния. Кальций и магний в щелочной среде образуют прочные комплексы с ЭДТА; этот реактив в первую очередь реагирует с кальцием. Если проводить реакцию при высоком значении рН раствора (12-13) и применять индикатор, вступающий в реакцию только с кальцием, можно определить его содержание в воде и почве.
При содержании в пробе больших количеств органических веществ проводят предварительную обработку (органические вещества удаляют сухим или мокрым сжиганием). Определению мешают ионы тяжелых металлов и железа.
Суммарная относительная погрешность, выраженная коэффициентом вариации для этого метода при определении кальция в шламе и почве, составляет, %: для количества эквивалентов кальция от 0,5 до 2 ммоль в 100 г почвы - 12,5; свыше 6 ммоль - 5.
Реже применяются следующие методы.
Перманганатометрический метод основан на осаждении кальция в виде оксалата, растворение этой соли в серной кислоте и определении количества щавелевой кислоты титрованием перманганатом.
Ацидиметрический метод основан на осаждении кальция в виде щавелевокислой его соли, которую затем прокаливают, получая при этом окись кальция в результате разложения оксалата. Образовавшуюся окись кальция растворяют в воде и оттитровывают кислотой. Существенным недостатком этих методов являются длительность их выполнения и необходимость проведения большого количества подготовленных трудоемких операций.
Сущность спектроскопического метода заключается в регистрации спектров исследуемого образца и сравнении их со спектрами эталонных растворов. Определения проводят с помощью спектрографа ИСП-22 в дуговом режиме генератора при силе тока 8 А и времени экспозиции 30 с. Метод очень чувствителен, точен, но требует специального приборного оснащения, а для приготовления эталонных растворов необходимы спектрально-чистые соли кальция.
Анализируя изложенные методы определения кальция, учитывая доступность используемых для их проведения приборов и реактивов, трудоемкость и длительность анализов, можно рекомендовать как наиболее приемлемый во всех отношениях метод комплексонометрического определения.
Магний. Природные воды содержат обычно меньше соединений магния, чем кальция, чего нельзя сказать о сточных водах, в которых возможно как присутствие соединений обоих металлов в любых соотношениях, так и наличие только одних соединений магния.
Для питьевых, подземных и поверхностных вод согласно ГОСТ 4151 — 72 определяется общая жесткость. Это определение проводится комплексонометрическим методом. Он основан на связывании ионов кальция и магния в комплексные соединения, сопровождающемся изменением окраски индикатора. Чувствительность этого метода зависит от чувствительности применяемых индикаторов и составляет 1 - 2 мкг-экв/л (для индикаторов кислотный хром синий марки К и кислотный хром темно-синий), при меньшей жесткости окраска комплексов индикаторов с ионами кальция и магния уже не отмечается визуально. Максимально возможная ошибка при титровании из обычной бюретки 0,1 м раствором трилона Б при общей жесткости 5 - 20 мг-экв/л составляет 1- 2%. Перед определением общей жесткости из пробы удаляют взвешенные и коллоидные вещества. Для сильноминерализованных проб следят, чтобы концентрация других ионов не превышала предельно допустимые концентрации мешающих ионов. Зная общую жесткость исследуемой пробы и содержание в ней кальция, содержание магния определяют по разности значений общей и кальциевой жесткости.
Содержание магния в воде при отсутствии солей кальция определяют титрованием иона магния комплексоном III в присутствии индикатора эриохром-черного или гравиметрическим методом, который основан на малой растворимости двойной фосфорнокислой соли магния — аммония, образующейся в аммиачных растворах в виде белого кристаллического осадка. При прокаливании этого вещества оно теряет воду и аммиак, превращаясь в пирофосфат магния, который и взвешивают. Иногда вместо весового окончания этого метода может быть рекомендовано ацидиметрическое. Этот метод имеет те же недостатки, что и весовой метод определения кальция, и еще в большей степени зависит от опыта аналитика.
Возможно комплексонометрическое определение кальция и магния при совместном присутствии в одной пробе. Сущность этого метода заключается в двойном последовательном комплексонометрическом титровании в одной пробе ионов кальция при рН = 12,54÷13 в присутствии индикатора мурексида и ионов магния при рН около 10 с эриохром-черным марки Т.
Сущность атомно-абсорбционного метода определения ионов, в том числе ионов магния, изложена выше. Для предотвращения образования в пламени труднодиссоциируемых соединений магния с сопутствующими элементами в анализируемую пробу вводят избыток стронция. Для определения магния используют аналитическую линию 285,2 нм. Из аналитической практики по определению состава и свойств отходов бурения известно, что магний присутствует в значительно меньших количествах, чем кальций. Поэтому целесообразно применять комплексонометрическое определение кальция и магния.
Железо. В небольших количествах железо присутствует почти во всех сточных водах. Формы содержания его в растворенном состоянии различны: коллоидный раствор вследствие пентизации гидррксида железа органическими веществами, комплексные соединения с неорганическими и органическими лигандами, суспензированные в воде твердые частицы. Кроме того, оно может быть в трехвалентном или двухвалентном состоянии. При рН раствора более 3,5 железо (III) присутствует в водной фазе в виде комплекса, а при рН более 8 железо (II) не существует в растворе в виде свободных ионов, не связанных в комплекс. Надо также учитывать то, что в воде, содержащей кислород, железо (П) легко переходит в железо (III) и осаждается в виде гидроксида. Железо (II) достаточно устойчиво в водном растворе только в сильнокислой среде.
Таким образом, точные результаты могут быть получены только при определении суммарного содержания железа во всех его формах. Раздельное определение растворенного и нерастворенного железа, а также железа (II) и железа (III) дает менее достоверные результаты.
Для определения общего содержания железа в различных водах предлагается атомно-абсорбционный метод и три колориметрических метода: с роданидом, сульфосалициловой кислотой и о-фенантролином и комплексонометрический метод.
Колориметрическое определение железа с роданидом основано на окислении всего содержащегося в пробе железа до трехвалентного, которое в кислой среде реагирует с роданидом, вызывая красное окрашивание. Интенсивность окраски пропорциональна концентрации железа. Прямо можно определять 0,05 — 5,0 мг железа в 1 л воды с точностью ± 0,05 мг. Определению мешают многие катионы, например, ионы меди, висмута, кобальта. Считают, что обычно они отсутствуют. Мешающее влияние высокого содержания органических веществ и трудно разлагаемых комплексов железа устраняют выпариванием пробы с азотной или серной кислотой.
Сущность фотометрического метода с о-фенантролином заключается в том, что ионы железа (II) образуют с о-фенантролином оранжево-красные комплексные ионы, в которых один ион железа (II) соединен с тремя молекулами о-фенантролина. Для определения общего содержания железа предварительно восстанавливают железо (III) гидрохлоридом гидроксиламина. Окраска не зависит от рН в границах от 3 до 9 и очень устойчива. Интенсивность окраски раствора пропорциональна концентрации железа. Прямое определение возможно при содержании железа 0,05 - 2,0 мг в 1 л воды. Мешают определению сильные окислители, нитриты, фосфаты, особенно полифосфаты, хром и цинк в концентрациях, превышающих концентрации железа более чем в 10 раз. Определению мешает медь в концентрациях, превышающих 10 мг/л. Влияние ее можно снизить при работе в области рН от 2,5 до 4,0. Мешающее влияние органических веществ и прочных комплексных соединений железа устраняют минерализацией пробы с азотной или серной кислотой.
Колориметрическое определение с сульфосалициловой кислотой основано на реакции сульфосалициловой кислоты с солями железа в щелочной среде с образованием желтого комплекса железа. Этим способом можно определить 0,1 — 10 мг/л железа. Точность определения ± 0,1 мг/л. Затруднения при определении создаются собственной окраской пробы; окраску уничтожают окислением или проводят холостой опыт, в котором окрашенную пробу обрабатывают так же, как и при анализе, но без сульфосалициловой кислоты и изменяют оптическую плотность по отношению к основной пробе. Определению железа мешают высокие концентрации алюминия и меди, которые, подобно железу, образуют цветные комплексы. В таких случаях выбирают другой метод определения.
Определение общего содержания железа производят колориметрически с применением о-фенантролина, сульфосалицилита натрия и роданида (ГОСТ 4011-72).
Комплексонометрический метод предназначен для определения больших концентраций железа. Сущность метода состоит в том, что железо, содержащееся в сточной воде, сначала окисляют до трехвалентного, затем осаждают аммиаком. Осадок растворяют в соляной кислоте и заканчивают определение титрованием ЭДТА с сульфосалициловой кислотой в качестве индикатора.
На практике для нахождения содержания железа пользуются одним из колориметрических методов определения с учетом имеющихся в лаборатории реактивов и содержащихся в пробе мешающих ионов.
Алюминий. В природных водах алюминий находится только в малых концентрациях, которые, как правило, не превышают десятых долей миллиграмма в 1 л воды, ему обычно сопутствует железо. Соли алюминия в воде гидролизуются и выпадают в виде осадка гидроокиси. В сильнокислой среде алюминий присутствует в виде катионов, в сильнощелочной — в виде алюминат-ионов. Содержание алюминия определяют в водах, которые подвергались очистке с применением алюминиевой соли, а также контролю работы станций водоподготовки. В сточных водах алюминий определяют только тогда, когда он является основной частью примесей.
Для определения алюминия, находящегося в
E:\Ellina\Научная и учебная литература\05_Глава 4_4.3.6.docx
E:\Ellina\Научная и учебная литература\05_Глава 4_4.3.6.docx
E:\Ellina\Научная и учебная литература\05_Глава 4_4.5.docx
E:\Ellina\Научная и учебная литература\06_Глава 5.docx
растворе, используется несколько методов. Один из них — атомно-абсорбционный.
Для определения алюминия, находящегося в растворе в малых концентрациях, существует большое число фотометрических методов. Большинство из них (например, методы с применением алюминона, ализаринсульфоната) основаны на образовании коллоидных окрашенных растворов (лаков) — соединений этих реагентов с алюминием. Поскольку как интенсивность получаемых окрасок, так и оттенки их зависят от степени дисперсности образующихся коллоидных частиц лаков, а последняя в свою очередь зависит от многих факторов (присутствия различных солей в растворе и их концентрации, температуры, скорости влияния реактива и т.д.), методы эти не рекомендуются для определения алюминия в промышленных сточных водах [173]. Для них рекомендуют два фотометрических метода, в которых анализируются истинные растворы окрашенных веществ.
Первый из них - метод с экстракцией гидроксихинолята алюминия. Метод основан на том, что гидроксихинолят количественно экстрагируется хлороформом в слабокислой среде (рН = 4,35÷4,5), окрашивая хлороформный слой в желто-зеленый цвет. Измерения проводят при длине волны 387 - 400 нм.
Экстрагирование хлороформом дает возможность определять алюминий в широком диапазоне содержания его в пробе. При обработке 100 мл пробы 10 мл хлороформа можно определять алюминий, в концентрациях более 0,01 мг/л.
Из элементов, мешающих определению, важнейший — железо. Его предварительно переводят в трехвалентную форму, окислив персульфатом аммония, и извлекают в виде оксихинолята железа (III) хлороформом из более кислой среды (рН = 1,8 ÷ 2,0), из которой оксихинолят алюминия не извлекается. При желании железо можно попутно колориметрически определять в хлороформном экстракте. Вместе с железом (III) извлекается и медь.
Извлекаемые хлороформом и окрашивающие его органические вещества удаляют предварительной экстракцией этим же растворителем.
Одновременно с алюминием в хлороформный экстракт переходят висмут, индий, никель и цинк (частично), однако эти элементы обычно в водах присутствуют в количествах, настолько малых по сравнению с содержанием в них алюминия, что влиянием их в большинстве случаев можно пренебречь.
Второй метод — фотометрический с применением эриохромцианина Р. Сущность метода заключается в том, что соли алюминия в разбавленных растворах при рН = 6,9 реагируют с эриохромцианином Р с образованием красно-фиолетового комплексного соединения, имеющего максимум светопоглощения при 535 нм. Оптимальное содержание алюминия составляет 20 - 30 мкг/л. Более концентрированные растворы предварительно разбавляют, более разбавленные подкисляют и упаривают. Определению мешают железо (III), марганец и магний в больших концентрациях, так как эти ионы образуют с эриохромцианином Р окрашенные соединения. Соли железа (III) в малых концентрациях восстанавливают солянокислым гидроксиламином.
Фосфаты, фториды и органические вещества понижают интенсивность окраски. Когда присутствует большое количество фосфатов, алюминий определяют другим способом. Органические вещества и фториды удаляют выпариванием с сильными кислотами.
Колориметрические методы определения алюминия с применением алюминона и стильбазо рекомендуются для количественного его определения в питьевых и поверхностных водах.
Хром. В качестве термостабилизирующей и ингибирующей добавки для сохранения подвижности буровых растворов при высоких забойных температурах используют хроматы и бихроматы щелочных металлов. Хотя добавки их не превышают десятых долей процента, оценивать содержание токсичного хрома в отходах бурения в некоторых случаях будет необходимо. Хром (VI) в щелочных растворах чаще всего находится в виде хромат-ионов. В присутствии восстановителей шестивалентный хром может перейти в трехвалентный. Поэтому обычно определяют общее содержание хрома в растворе или твердой фазе в зависимости от цели анализа. В справочной литературе для анализа хрома в воде рекомендуются титриметрический метод определения хрома (VI) с сульфатом железа (II) и колориметрический метод определения с дифенилкарбазидом. Этими же методами определяют и общее содержание хрома в пробе. Содержание хрома (III) устанавливают по разности результатов определения общего и шестивалентного хрома.
Если анализируемая проба имеет щелочную реакцию или в ней содержатся в больших количествах органические вещества или хлориды, предлагается фотометрический метод определения малых количеств хрома [99]. Этот метод пригоден для анализа наиболее сложных по составу вод. Сущность метода сводится к следующему. В одной порции пробы определяют содержание хрома (III), для чего его осаждают оксидом магния при рН — 10,5÷11,0. Осадок гидроксида хрома сорбируется на оксиде магния, который отфильтровывают, затем растворяют в серной кислоте и окисляют хром (III) до хрома (VI) персульфатом аммония или прокаливают со смесью карбоната натрия и оксида магния, в результате чего хром (III) также окисляется до хрома (IV). Заканчивают анализ фотометрическим определением с дифенилкарбазидом. В другой порции испытуемой воды определяют суммарное содержание хрома (III) и хрома (IV), для чего сначала восстанавливают хром (VI) до хрома (III) сернистой кислотой, затем осаждают хром (III) оксидом магния и определяют его, как в первой порции.
Хром в любых водах может быть определен атомно-абсорбционным методом, который подробно был описан ранее. Этот метод требует специального оборудования.
Несмотря на результаты исследований [212], установивших, что большинство ОБР не содержит опасных для окружающей среды и человека концентраций тяжелых металлов, на наш взгляд, необходимо определение не только ионов бария и хрома в отходах бурения, но и олова, цинка, свинца, меди. Наибольшей чувствительностью на эти компоненты обладают атомно-абсорбционная спектрометрия и адсорбционная полярография.
Исходя из обобщения отечественного и зарубежного опыта проведения физико-химических анализов БСВ, ОБР и БШ и учитывая требования контролирующих природоохранных органов, наиболее эффективными, надежными и приемлемыми, обеспечивающими необходимую для практики степень точности, являются следующие методы: весовой и ИК-спектрометрический для определения нефти и нефтепродуктов; титриметрические и фотоэлектроколори-метрические для определения анионного состава стоков; пламенно-эмиссионный и атомно-абсорбционный для определения катионов идентификации.
На такие методы должны ориентироваться производственные службы предприятий нефтегазовой промышленности, а также проектные институты и промысловые лаборатории производственных объединений, занимающиеся охраной окружающей среды и проведением контроля за ее состоянием.
Дальнейшее развитие методов и техники анализа производственно-технологических отходов бурения во многом будет определяться требованиями природоохранных органов, предъявляемыми к точности анализа в соответствии с действующими нормативами (ПДК, ПДС, ПДВ и т.д.), а также приборной (инструментальной) базой и совершенствованием известных методик проведения таких работ.

