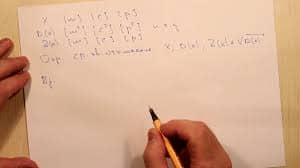2015-05-06
2015-05-06 6701
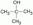
6701ArHal + СH3(CH2)nCH2Hal + 2Na ® Ar-CH2(CH2)nCH3 + 2NaHal Относится к реакциям кросс-сочетания. Наил.результаты достиг. при исп.арилбромидов и первичных алкилбромидов.
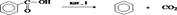
23. Хим. св-ва аренов. Р-и электрофильного замещения в молекуле бензола. I.Реакции замещения
1.Взаимодействие с галогенами 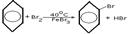
2.Взаимодействие с галогензамещенными алканами 
3.Взаимодействие с непредельными углеводородами 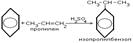
4.Реакция нитрования
II.Реакции присое 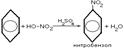 динения
динения
1.Присоединение водорода 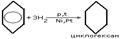
2.Присоединение хлора на свету 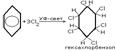
В реакциях электрофил. замещ. атакующей частицей явл. катионы или мол-лы с вакантной орбиталью, а также мол-лы с сильно поляризованной связью, кот. разрывается гетеролитически в процессе р-и. Как пр., электрофил. ч-цы обр. в реакционной смеси. Уходящая группа отщепляется, оставляя субстрату свою пару электронов и должна быть слабой кислотой Льюиса. В р. ароматич. электрофил. замещения такой группой чаще всего служит протон.

Механизм ароматического электрофильного замещения.
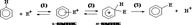
Реакция нач. с обр. донорно-акцеп. комплекса между ареном и электрофилом (p-комплекс), в кот. арен выступает в роли p-основания. Далее p-комплекс перегруппировывается в s-комплекс, содержащий s-связь углерод-электрофил. При образовании аренониевого иона ароматическая p-с-ма нарушается и образуется незамк. сопряженная с-ма циклогексадиенильного катиона, в кот. атом углерода, образующий связь с электрофилом, нах. в состоянии sp3-гибридизации, а положит. заряд делокализован по сопряженной с-ме. Делокализация положит. заряда происх. в основном за счет орто- и пара-положений по отношению к вступающему электрофилу.
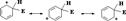
Аренониевый ион является высокореакционноспособной ч-цей, кот. может стабилизироваться либо за счет присоединения нуклеофила, как это происходит при электрофильном присоединении к алкенам, либо путем отщепления протона под действием основания – энергетич. более выгодно: приводит к восстановлению ароматической с-мы. Именно оно и реализуется при ароматическом электрофильном замещении.Для многих реакций ароматического электрофильного замещения наблюдается корреляция между устойчивостью σ-комплекса и скоростью реакции. Это означает, что скорость определяется стадией образования s-комплекса и s-комплекс является хорошей моделью переходного состояния.
При смешивании аренов с электрофилами образуются слабые p-комплексы.
24. Хим св-ва аренов. Р. электрофиль замещения. Ароматическими углеводородами (аренами) называются вещества, в молекулах которых содержится одно или несколько бензольных колец — циклических групп атомов углерода с особым характером связей. Понятие “бензольное кольцо” требует расшифровки. Для этого необходимо рассмотреть строение молекулы бензола. Первая структура бензола была предложена в 1865г. немецким ученым А. Кекуле:
Эта формула правильно отражает равноценность шести атомов углерода, однако не объясняет ряд особых свойств бензола. Например, несмотря на ненасыщенность, бензол не проявляет склонности к реакциям присоединения: он не обесцвечивает бромную воду и раствор перманганата калия, т. е. не дает типичных для непредельных соединений качественных реакций.Бензольное ядро облад высокой прочностью, чем объясняется склонность к р-ям замещ. В отличие от алканов, кот также склонны к р-ям замещ, арены хар-тся большой подвижностью атомов водорода в ядре, поэтому р-ии галогенирования, нитрования, сульфирования и др. протекают в значительно более мягких условиях,чем у алканов. Обладая подвижной шестеркой p -электронов, ароматическое ядро является удобным объектом для атаки электрофильными реагентами. Этому способствует также пространственное расположение p -электронного облака с двух сторон плоского s -скелета молекулы.Для аренов наиболее характерны реакции, протекающие по механизму электрофильного замещения, обозначаемого символом-SE 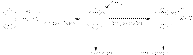 Механизм электрофильного замещения можно представить следующим образом. Электрофильный реагент XY (X является электрофилом) атакует электронное облако, и за счет слабого электростатического взаимодействия образуется неустойчивый p -комплекс. Ароматическая система при этом еще не нарушается. Эта стадия протекает быстро. На второй, более медленной стадии формируется ковалентная связь между электрофилом Х и одним из атомов углерода кольца за счет двух p -электронов кольца. Этот атом углерода переходит из sр2 - в sр3-гибридное состояние. Ароматичность системы при этом нарушается. Четыре оставшиеся p -электрона распределяются между пятью другими атомами углерода, и молекула бензола образует карбокатион, или s -комплекс. Нарушение ароматичности энергетически невыгодно, поэтому структура s -комплекса менее устойчива, чем ароматическая структура. Для восстановления ароматичности происходит отщепление протона от атома углерода, связанного с электрофилом (третья стадия). При этом два электрона возвращаются в p -систему и тем самым восстанавливается арома
Механизм электрофильного замещения можно представить следующим образом. Электрофильный реагент XY (X является электрофилом) атакует электронное облако, и за счет слабого электростатического взаимодействия образуется неустойчивый p -комплекс. Ароматическая система при этом еще не нарушается. Эта стадия протекает быстро. На второй, более медленной стадии формируется ковалентная связь между электрофилом Х и одним из атомов углерода кольца за счет двух p -электронов кольца. Этот атом углерода переходит из sр2 - в sр3-гибридное состояние. Ароматичность системы при этом нарушается. Четыре оставшиеся p -электрона распределяются между пятью другими атомами углерода, и молекула бензола образует карбокатион, или s -комплекс. Нарушение ароматичности энергетически невыгодно, поэтому структура s -комплекса менее устойчива, чем ароматическая структура. Для восстановления ароматичности происходит отщепление протона от атома углерода, связанного с электрофилом (третья стадия). При этом два электрона возвращаются в p -систему и тем самым восстанавливается арома 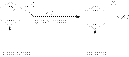 тичность:
тичность:
Реакции электрофильного замещения широко используются для синтеза многих производных бензола. Галогенирование. При взаимод бензола с галогеном атом водорода ядра замещается галогеном. С6Н6+Сl2=С6Н5Сl+HCl. Нитрование. При действии на бензол нитрующей смеси атом водорода замещается нитрогруппой (нитрующая смесь – это смесь концентрированных азотной и серной кислот в соотношении 1:2 соответственно). С6Н6+HNO3=C6H5NO2 нитробензол+H2O. Сульфирование осущ конц H2SO4 или олеумом. В процессе р-ии водородный атом замещается сульфогруппой. C6H6 + H2SO4 —SO3à C6H5 – SO3Hбензосульфокислота + H2O. ) Алкилирование. Замещение атома водорода в бензольном кольце на алкильную группу (алкилирование) происходит под действием алкилгалогенидов (реакция Фриделя-Крафтса) или алкенов в присутствии катализаторов AlCl3, AlBr3, FeCl3 (кислот Льюиса).С6Н6+СН3Cl=C6H5CH3+HCl. Гидрирование. Присоединение водорода осущ только в присутствии катализаторов и при повыш t. Бензол гидрируется с образованием циклогексана, а производные бензола дают производные циклогексана.C6H6+3H2=C6H12. Галогенирование. Радикальное хлорирование. В условиях радикальных р-ий (ультрафиолетовый свет, повыш t) возможно присоединение галогенов к аромат соед-ям. При радикальном хлорировании бензола получен "гексахлоран" (средство борьбы с вредными насекомыми). C6H6+3Cl2=C6H6Cl6.К реакции электрофильного замещения относятся реакции Фриделя-Крафтса (т.е. алкилирование и ацилирование аромат ядра в присутствии AlCl3). Взаимод с н-электрофилами. Сильн к-ты с аренами образ z-комплекс и происход образование бензониевого иона. Происходит обмен атомов Н, а в присутствии дейтерированных к-т-дейтерообмен.
25 Хим св-ва аренов. Гомологи бензола. Присоед. Ароматическими углеводородами (аренами) называются вещества, в молекулах которых содержится одно или несколько бензольных колец — циклических групп атомов углерода с особым характером связей. Понятие “бензольное кольцо” требует расшифровки. Для этого необходимо рассмотреть строение молекулы бензола. Первая структура бензола была предложена в 1865г. немецким ученым А. Кекуле: Обладая подвижной шестеркой p -электронов, ароматическое ядро является удобным объектом для атаки электрофильными реагентами. Этому способствует также пространственное расположение p -электронного облака с двух сторон плоского s -скелета молекулы. Реакции в боковой цепи. По химическим свойствам алкильные радикалы подобны алканам. Атомы водорода в них замещаются на галоген по свободно-радикальному механизму. Поэтому в отсутствие катализатора при нагревании или УФ-облучении идет радикальная реакция замещения в боковой цепи. Влияние бензольного кольца на алкильные заместители приводит к тому, что замещается всегда атом водорода у атома углерода, непосредственно связанного с бензольным кольцом (a -атома углерода).  Замещение в бензольном кольце возможно только по механизму SE в присутствии катализатора АlСl3:
Замещение в бензольном кольце возможно только по механизму SE в присутствии катализатора АlСl3:  При действии на гомологи бензола перманганата калия и других сильных окислителей боковые цепи окисляются. Какой бы сложной ни была цепь заместителя, она разрушается, за исключением a -атома углерода, который окисляется в карбоксильную группу.Гомологи бензола с одной боковой цепью дают бензойную кислоту:
При действии на гомологи бензола перманганата калия и других сильных окислителей боковые цепи окисляются. Какой бы сложной ни была цепь заместителя, она разрушается, за исключением a -атома углерода, который окисляется в карбоксильную группу.Гомологи бензола с одной боковой цепью дают бензойную кислоту:  СЕЛЕКТИВНОСТЬ.Основная реакция изомеризации сопровождается побочными реакциями - расщепления, уплотнения и др., особенно при изомеризации пентана и гексана. Для подавления побочных реакций процесс ведется в присутствии небольших количеств водорода. Помимо этих основных реакций изомеризации, элкилнафтеновые углеводороды, а также перафины могут подвергаться гидрокрекингу, в результате чего появляются низкомолекулярные парафины и изонара-финн. Поскольку термодинамические расчеты показывают, что помимо основной реакции изомеризации могут протекать реакции диспропорционирования и гидрирования ароматических углеводородов, для повышения селективности процесса необходимы катализаторы, в присутствии которых побочные реакции протекают с минимальными скоростями. Например, в присутствии катализаторов, интенсифицирующих реакции диспропорционирования, в сырье целесообразно добавлять толуол. При проведении процесса под давлением водорода добавление в сырье нафтеновых углеводородов С8 может предотвратить их образование. Поскольку термодинамические расчеты показывают, что помимо основной реакции изомеризации могут протекать реакции диспро-порайонирования и гидрирования ароматических углеводородов, для повышения селективности процесса необходимы катализаторы, в присутствии которых побочные реакции протекают с минимальными скоростями. Можно также добавлять в сырье продукты превращения ароматических углеводородов, чтобы сдвинуть равновесие в сторону образования ароматических углеводородов С8, Например, в присутствии катализаторов, интенсифицирующих реакции диспропор-ционирования, в сырье целесообразно добавлять толуол. При проведении процесса под давлением водорода добавление в сырье нафте - новых углеводородов С8 может предотвратить их образование. Реакции присоединения водорода
СЕЛЕКТИВНОСТЬ.Основная реакция изомеризации сопровождается побочными реакциями - расщепления, уплотнения и др., особенно при изомеризации пентана и гексана. Для подавления побочных реакций процесс ведется в присутствии небольших количеств водорода. Помимо этих основных реакций изомеризации, элкилнафтеновые углеводороды, а также перафины могут подвергаться гидрокрекингу, в результате чего появляются низкомолекулярные парафины и изонара-финн. Поскольку термодинамические расчеты показывают, что помимо основной реакции изомеризации могут протекать реакции диспропорционирования и гидрирования ароматических углеводородов, для повышения селективности процесса необходимы катализаторы, в присутствии которых побочные реакции протекают с минимальными скоростями. Например, в присутствии катализаторов, интенсифицирующих реакции диспропорционирования, в сырье целесообразно добавлять толуол. При проведении процесса под давлением водорода добавление в сырье нафтеновых углеводородов С8 может предотвратить их образование. Поскольку термодинамические расчеты показывают, что помимо основной реакции изомеризации могут протекать реакции диспро-порайонирования и гидрирования ароматических углеводородов, для повышения селективности процесса необходимы катализаторы, в присутствии которых побочные реакции протекают с минимальными скоростями. Можно также добавлять в сырье продукты превращения ароматических углеводородов, чтобы сдвинуть равновесие в сторону образования ароматических углеводородов С8, Например, в присутствии катализаторов, интенсифицирующих реакции диспропор-ционирования, в сырье целесообразно добавлять толуол. При проведении процесса под давлением водорода добавление в сырье нафте - новых углеводородов С8 может предотвратить их образование. Реакции присоединения водорода 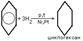
Присоединение хлора на свету
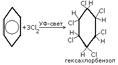


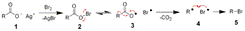 26Получение галогеналканов. Прямое галогенирование. Радикальным галогенированием можно получать хлор- или бромалканы. Недостаток метода: образ-ие смеси различных продуктов замещения. При этом наряду с изомерными монозамещёнными в смеси также содержатся ди- и полизамещённые соединения. К тому же, напр, эквимолярная смесь Cl2 и CH4 взрывоопасна, не говоря уже о смесях алканов со фтором. Однако меняя условия процесса можно добиться приемлемых для промышленности выходов. Напр, при хлорировании алкан берут в избытке. Продукты разделяют фракционной перегонкой. Так в промышленности получают метиленхлорид и тетрахлоруглерод.Алканы хлорируются при интенсивном УФ-облучении или нагревании. Наиболее легко образуются третичные радикалы и, соотв-но, галогеналканы; наименее – первичные. Бромирование мало характерно для алканов легче гексана, а прочие бромируются при одновременном освещении и кипячении.
26Получение галогеналканов. Прямое галогенирование. Радикальным галогенированием можно получать хлор- или бромалканы. Недостаток метода: образ-ие смеси различных продуктов замещения. При этом наряду с изомерными монозамещёнными в смеси также содержатся ди- и полизамещённые соединения. К тому же, напр, эквимолярная смесь Cl2 и CH4 взрывоопасна, не говоря уже о смесях алканов со фтором. Однако меняя условия процесса можно добиться приемлемых для промышленности выходов. Напр, при хлорировании алкан берут в избытке. Продукты разделяют фракционной перегонкой. Так в промышленности получают метиленхлорид и тетрахлоруглерод.Алканы хлорируются при интенсивном УФ-облучении или нагревании. Наиболее легко образуются третичные радикалы и, соотв-но, галогеналканы; наименее – первичные. Бромирование мало характерно для алканов легче гексана, а прочие бромируются при одновременном освещении и кипячении. 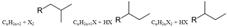 Или так написать CnH2n+2 + X2
Или так написать CnH2n+2 + X2 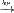 CnH2n+1X + HX
CnH2n+1X + HX 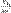 CnH2nX2 + HX
CnH2nX2 + HX 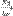 ... В присутствии специальных реагентов, а также инициаторов свободных радикалов замещённые алкены бромируются в аллильное положение. или так писать
... В присутствии специальных реагентов, а также инициаторов свободных радикалов замещённые алкены бромируются в аллильное положение. или так писать 
 Аллильное хлорирование осуществляется лишь при 400–600°C, обычно же идут конкурирующие реакции – присоединение по двойной связи, полимеризация, изомеризация алкенов. В промышленности так производят аллилхлорид.Замещённые арены хлорируются и бромируются в боковую цепь. Например, толуол при нагревании и интенсивном освещении хлорируют, получая бензилхлорид.Радикальным методом получают также и перфторалканы. Реакция протекает очень энергично, и вследствие большого тепловыделения приходится разбавлять фтор азотом, применяют также медные сетки для отвода теплоты. В процессе применяют переносчики фтора – фториды металлов, как CoF2, MnF2, AgF, образующие в ходе реакции соответственно CoF3, MnF4, AgF2. Получение моногалогеналканов Присоединение галогеноводородов к алкенам. R-CH=CH2+HCl→R-CHCl-CH3. Весьма легко присоединяется фтор, иод присоединяется медленно. Обычно происходит стереоселективно, кроме как в присутствии свободных радикалов. В присутствии других нуклеофилов возможно сопряжённое присоединение.Реакции спиртов с галогеноводородами.
Аллильное хлорирование осуществляется лишь при 400–600°C, обычно же идут конкурирующие реакции – присоединение по двойной связи, полимеризация, изомеризация алкенов. В промышленности так производят аллилхлорид.Замещённые арены хлорируются и бромируются в боковую цепь. Например, толуол при нагревании и интенсивном освещении хлорируют, получая бензилхлорид.Радикальным методом получают также и перфторалканы. Реакция протекает очень энергично, и вследствие большого тепловыделения приходится разбавлять фтор азотом, применяют также медные сетки для отвода теплоты. В процессе применяют переносчики фтора – фториды металлов, как CoF2, MnF2, AgF, образующие в ходе реакции соответственно CoF3, MnF4, AgF2. Получение моногалогеналканов Присоединение галогеноводородов к алкенам. R-CH=CH2+HCl→R-CHCl-CH3. Весьма легко присоединяется фтор, иод присоединяется медленно. Обычно происходит стереоселективно, кроме как в присутствии свободных радикалов. В присутствии других нуклеофилов возможно сопряжённое присоединение.Реакции спиртов с галогеноводородами.
R-OH+H-Cl→R-Cl+H2O. Взаимодействие галогенидов фосфора или тионилхлорида со спиртами.
3R-OH+PCl3→3R-Cl+H3PO3. Реакция Бородина — Хунсдикера 
Реакция Сварта R-Cl+AgF→R-F+AgCl. Получение дигалогеналканов Присоединение галогеноводородов к алкинам.
R-C≡CH+2HCl→R-CCl2-CH3. Взаимодействие альдегидов и кетонов с PCl5, PBr5 или SF4. Рекция идёт при нагревании.
R—CHO + PCl5 → R—CHCl2, Присоединение галогенов к алкенам R-CH=CH2 + Cl2 → R-CHCl-CHCl Раскрытие циклических простых эфиров при реакции с NaI в среде H3PO4+P2O5.
C4H8O + HI → I-CH2CH2CH2CH2-I При 180 °C ТГФ с хлороводородом даёт 1,4-дихлорбутан. Получение галогеналкенов. С алкинами напрямую — как H-электрофилы галогеноводороды взаимодействуют медленно. Реакцию значительно ускоряют соли меди и ртути; по сути, происходит нуклеофильное присоединение галогенид-иона. Например, есть такой способ производства винилхлорида в газовой фазе на активированном угле и сулеме при 120—180 °C.Также и галогены присоединяются по тройной связи медленнее, чем по двойной. Вероятно, реакция идёт через КПЗ, который опять-таки является уже электрофильной частицй.Или применяют производные алкенов. В частности, для получения того же винилхлорида есть такие два способа.Дегидратация этиленхлоргидрина на катализаторе.Дегидрохлорирование дихлорэтана в газовой фазе при 400—500 °C. Получение галогенаренов Галогенирование аренов или алкиларенов в ядро
Ph-H+Cl2→Ph-Cl Разложение арендиазониевых солей Ph-N≡N Cl→Ph-Cl Получение бензилгалогенидов Галогенирование алкиларенов в боковую цепь. Ph-CH3+Cl2→Ph-CH2Cl Хлорметилирование(р-ция Блана)
Ph-H+HCHO+HCl→Ph-CH2Cl
 27Хими св-ва галогеналканов. Взаимодействие галогеналканов с металлами.Взаимодействие с металлами 1--Реакция Вюрца (взаимодействие галогенпроизводных алканов с Na) CH3Cl + 2Na + ClCH3 -> 2NaCl + CH3 - CH3 -\\- 2C2H5Cl + 2Na -> 2 Nacl + C4H10 ------ CH3Cl + 2Na + C2H5Cl -> 2 NaCl + C3H8 -----------2--Гидролиз р-ва Гриньяра H2O -----RX+Mg→RMgX→RH. Р-в Гриньяра Пример Mg H2O
27Хими св-ва галогеналканов. Взаимодействие галогеналканов с металлами.Взаимодействие с металлами 1--Реакция Вюрца (взаимодействие галогенпроизводных алканов с Na) CH3Cl + 2Na + ClCH3 -> 2NaCl + CH3 - CH3 -\\- 2C2H5Cl + 2Na -> 2 Nacl + C4H10 ------ CH3Cl + 2Na + C2H5Cl -> 2 NaCl + C3H8 -----------2--Гидролиз р-ва Гриньяра H2O -----RX+Mg→RMgX→RH. Р-в Гриньяра Пример Mg H2O 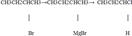
Бромистый втор-бутил Втор-бутил магний бромид н-бутан. 3--Восстановление металлом в кислоте RX+Zn+H+→RH+Zn+X−
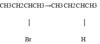 Бромистый втор-бутил н-бутил. 1. Галогеналканы легко вступают в реакции замещения. Это связано с тем, что связь С-Г полярна и на атоме углерода имеется частичный положительный заряд (+d). Поэтому атом углерода легко атакуется нуклеофилами, такими, как анионы ОН- и CN-, или соединениями с неподеленными электронными парами: NH3 и Н2О:. Галогенид-ион является, как говорят, «хорошей уходящей группой», поэтому реакции замещения идут достаточно легко:
Бромистый втор-бутил н-бутил. 1. Галогеналканы легко вступают в реакции замещения. Это связано с тем, что связь С-Г полярна и на атоме углерода имеется частичный положительный заряд (+d). Поэтому атом углерода легко атакуется нуклеофилами, такими, как анионы ОН- и CN-, или соединениями с неподеленными электронными парами: NH3 и Н2О:. Галогенид-ион является, как говорят, «хорошей уходящей группой», поэтому реакции замещения идут достаточно легко: 
Примерами таких реакций служит превращение галогеналканов в спирты и амины:

Условия, необходимые для протекания таких реакции, сильно зависят от строения галогеналкана и природы нуклеофила. В некоторых случаях достаточно простого смешения реагентов, в других — требуется длительное нагревание.
Легкость замещения атома галогена в значительной степени зависит и от природы галогена. Энергия связи ОТ увеличивается в ряду С—I<С—Br<С—Сl<С—F. В этом же ряду уменьшается реакционная способность галогеналканов. Связь С—F настолько прочна, что фторалканы практически не вступают в реакции нуклеофильного замещения. Активность остальных галогеналканов по отношению к нуклеофилам падает в ряду RI>RBr>RCl.
2. Галогеналканы также легко вступают в реакции отщепления. При этом образуются галогеноводород и алкен. Эти реакции протекают при действии основания на галогеналкан. Таким образом, при действии сильных оснований на Галогеналканы отщепляются молекулы НГ и образуются алкены:
RСН2СН2Г+ОН-®RCH2CH2OH+Г- — замещение RСН2СН2Г+ОН-®RCH=CH2+Н2O+Г- — отщепление (СН3)3ССl+ОН-®(СН3)2C-СН2+Н2O+Сl-
Скорости замещения и отщепления зависят также от структуры углеводородного радикала. Роль реакции замещения возрастает в следующем ряду галогеналканов:
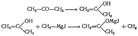
Первичные < Вторичные < Третичные. Третичные Галогеналканы вступают в реакцию замещения особенно легко (в отсутствие сильного основания):. (СН3)3ССl+Н2O®водный этанол,25° (СН3)3СОН+НСl В целом в присутствии водного раствора основания для первичных галогеналканов характерны в основном реакции отщепления, вторичные Галогеналканы дают, как правило, смесь продуктов отщепления и замещения, а третичные образуют главным образом продукты замещения. Галогеналканы вступают в SN-реакции с различными нуклеофилами, такими, как цианид-анион CN-, ацетат-анион СН3СОO-, аммиак:NH3, амины RN..H2 и многие другие. 3. При добавлении раствора галогеналкана в диэтиловом эфире СН3СН2ОСН2СН3 к магниевой стружке происходит экзотермическая реакция: магний переходит в раствор и образуется реактив Гриньяра формулы R—Mg—Г, где R — алкильная или арильная группа, а Г — галоген.
RГ+Mg®эфир R-Mg-Г. Реактивы Гриньяра вступают в реакции со многими соединениями, что позволяет использовать их в синтезе самых различных веществ. Синтез реактива Гриньяра R-X+Mg→RMgX Гриньяром было показано, что в присутствии абсолютного эфира галоидопроизводные углеводородов как жирного, так и ароматического ряда реагируют с металлическим магнием, образуя галоидомагнийорганические соединения (обычно называемые просто магнийорганическими соединениями):R-Hal + Mg R-MgHal Эти соединения отличаются большой химической активностью и способны вступать в разнообразные реакции. Водой магнийорганические соединения разлагаются с образованием соответствующих углеводородов:R-MgHal + H2O R-H + Mg(OH)Hal. Реакции магнийорганических соединений играют исключительно большую роль в органической химии, так как они могут быть использованы для синтеза самых разнообразных соединений.
28.Хим сво-ва галогеналканов. Нуклеофил замещение (SNu). Механизм реакций замещения SNu1 и SNu2 1. Галогеналканы легко вступают в реакции замещения. Это связано с тем, что связь С-Г полярна и на атоме углерода имеется частичный положительный заряд (+d). Поэтому атом углерода легко атакуется нуклеофилами, такими, как анионы ОН- и CN-, или соединениями с неподеленными электронными парами: NH3 и Н2О:Примерами таких реакций служит превращение галогеналканов в спирты и амины: Условия, необходимые для протекания таких реакции, сильно зависят от строения галогеналкана и природы нуклеофила. В некоторых случаях достаточно простого смешения реагентов, в других — требуется длительное нагревание. Легкость замещения атома галогена в значительной степени зависит и от природы галогена. Энергия связи ОТ увеличивается в ряду С—I<С—Br<С—Сl<С—F. В этом же ряду уменьшается реакционная способность галогеналканов. Связь С—F настолько прочна, что фторалканы практически не вступают в реакции нуклеофильного замещения. Активность остальных галогеналканов по отношению к нуклеофилам падает в ряду RI>RBr>RCl.
2. Галогеналканы также легко вступают в реакции отщепления. При этом образуются галогеноводород и алкен. Эти реакции протекают при действии основания на галогеналкан. RСН2СН2Г+ОН-®RCH2CH2OH+Г- — замещение RСН2СН2Г+ОН-®RCH=CH2+Н2O+Г- — отщепление (СН3)3ССl+ОН-®(СН3)2C-СН2+Н2O+Сl- Скорости замещения и отщепления зависят также от структуры углеводородного радикала. Роль реакции замещения возрастает в следующем ряду галогеналканов: Первичные < Вторичные < Третичные Третичные Галогеналканы вступают в реакцию замещения особенно легко (в отсутствие сильного основания): (СН3)3ССl+Н2O®водный этанол,25°(СН3)3СОН+НСl В целом в присутствии водного раствора основания для первичных галогеналканов характерны в основном реакции отщепления, вторичные Галогеналканы - отщепления и замещения, а третичные образуют главным образом продукты замещения. Галогеналканы вступают в SN-реакции с различными нуклеофилами, такими, как цианид-анион CN-, ацетат-анион СН3СОO-, аммиак:NH3, амины RN..H2 и многие другие. 3. При добавлении раствора галогеналкана в диэтиловом эфире СН3СН2ОСН2СН3 к магниевой стружке происходит экзотермическая реакция: магний переходит в раствор и образуется реактив Гриньяра формулы R—Mg—Г, где R — алкильная или арильная группа, а Г — галоген. RГ+Mg®эфир R-Mg-Г Реактивы Гриньяра вступают в реакции со многими соединениями, что позволяет использовать их в синтезе самых различных веществ. Реакции нуклеофильного замещения — реакции замещения, в которых атаку осуществляет нуклеофил — реагент, несущий неподеленную электронную пару. Уходящая группа в реакциях нуклеофильного замещения называется нуклеофуг. Все нуклеофилы являются основаниями Льюиса.Общий вид реакций нуклеофильного замещения: R−X + Y− → R−Y + X− (анионный нуклеофил)R−X + Y−Z → R−Y + X−Z (нейтральный нуклеофил) Выделяют реакции алифатического (широко распространены) и ароматического (мало распространены) нуклеофильного замещения. Реакции алифатического нуклеофильного замещения играют крайне важную роль в органическом синтезе и широко используются как в лабораторной практике, так и промышленности. Реакции SN1 Механизм реакции SN1 или реакции мономолекулярного нуклеофильного замещения (англ. substitution nucleophilic unimolecular) включает следующие стадии: 1. Ионизация субстрата с образованием карбкатиона (медленная стадия): R−X → R+ + X− 2. Нуклеофильная атака карбкатиона (быстрая стадия): R+ + Y− → R−Y или (если в качестве нуклеофила выступает нейтральная частица): R+ + Y−Z → R−Y+−Z 3. Отщепление катиона (быстрая стадия): R−Y+−Z → R−Y + Z+ Примером реакци  и SN1 является гидролиз трет-бутилбромида:Скорость реакции SN1 (в упрощённом виде) не зависит от концентрации нуклеофила и прямо пропорциональна концентрации субстрата[4]: Скорость реакции = k × [RX] Так как в процессе реакции образуется карбкатион, его атака (в идеальных условиях без учёта фактора влияния заместителей) нуклеофилом может происходить с обеих сторон, что приводит к рацемизации образующегося продукта. Важно иметь в виду, что SN1 механизм реализуется только в случае относительной устойчивости промежуточного карбкатиона, поэтому по такому пути, обычно, реагируют только третичные ((R)3C-X) и вторичные ((R)2CH-X) алкилпроизводные. Реакции SN2 Механизм реакции SN2 или реакции бимолекулярного нуклеофильного замещения (англ. substitution nucleophilic bimolecular) происходит в одну стадию, без промежуточного образования интермедиата. При этом атака нуклеофила и отщепление уходящей группы происходит одновременно: R−X + Y− → [Y⋯R⋯X]− → R−Y + X− Примером реакции SN2 является гидролиз этилбромида:
и SN1 является гидролиз трет-бутилбромида:Скорость реакции SN1 (в упрощённом виде) не зависит от концентрации нуклеофила и прямо пропорциональна концентрации субстрата[4]: Скорость реакции = k × [RX] Так как в процессе реакции образуется карбкатион, его атака (в идеальных условиях без учёта фактора влияния заместителей) нуклеофилом может происходить с обеих сторон, что приводит к рацемизации образующегося продукта. Важно иметь в виду, что SN1 механизм реализуется только в случае относительной устойчивости промежуточного карбкатиона, поэтому по такому пути, обычно, реагируют только третичные ((R)3C-X) и вторичные ((R)2CH-X) алкилпроизводные. Реакции SN2 Механизм реакции SN2 или реакции бимолекулярного нуклеофильного замещения (англ. substitution nucleophilic bimolecular) происходит в одну стадию, без промежуточного образования интермедиата. При этом атака нуклеофила и отщепление уходящей группы происходит одновременно: R−X + Y− → [Y⋯R⋯X]− → R−Y + X− Примером реакции SN2 является гидролиз этилбромида:  Условный энергетический профиль реакции бимолекулярного нуклеофильного замещения представлен на диаграмме Скорость реакции SN2 зависит как от концентрации нуклеофила, так и концентрации субстрата Скорость реакции = k × [RX] × [Y] Так как в процессе реакции атака нуклеофилом может происходить только с одной стороны, результатом реакции является стереохимическая инверсия образующегося продукта.
Условный энергетический профиль реакции бимолекулярного нуклеофильного замещения представлен на диаграмме Скорость реакции SN2 зависит как от концентрации нуклеофила, так и концентрации субстрата Скорость реакции = k × [RX] × [Y] Так как в процессе реакции атака нуклеофилом может происходить только с одной стороны, результатом реакции является стереохимическая инверсия образующегося продукта.
29. Хим св-ва галогеналканов. Реакция элеменирования. Механизм ЕNu1 и ЕNu2. Правило Зайцева. Химические свойства галогеналканов
1. Галогеналканы легко вступают в реакции замещения. Это связано с тем, что связь С-Г полярна и на атоме углерода имеется частичный положительный заряд (+d). Поэтому атом углерода легко атакуется нуклеофилами, такими, как анионы ОН- и CN-, или соединениями с неподеленными электронными парами: NH3 и Н2О:Примерами таких реакций служит превращение галогеналканов в спирты и амины:Условия, необходимые для протекания таких реакции, сильно зависят от строения галогеналкана и природы нуклеофила. В некоторых случаях достаточно простого смешения реагентов, в других — требуется длительное нагревание.
Легкость замещения атома галогена в значительной степени зависит и от природы галогена. Энергия связи ОТ увеличивается в ряду С—I<С—Br<С—Сl<С—F. В этом же ряду уменьшается реакционная способность галогеналканов. Связь С—F настолько прочна, что фторалканы практически не вступают в реакции нуклеофильного замещения. Активность остальных галогеналканов по отношению к нуклеофилам падает в ряду RI>RBr>RCl.2. Галогеналканы также легко вступают в реакции отщепления. При этом образуются галогеноводород и алкен. Эти реакции протекают при действии основания на галогеналкан. RСН2СН2Г+ОН-®RCH2CH2OH+Г- — замещение RСН2СН2Г+ОН-®RCH=CH2+Н2O+Г- — отщепление (СН3)3ССl+ОН-®(СН3)2C-СН2+Н2O+Сl- Скорости замещения и отщепления зависят также от структуры углеводородного радикала. Роль реакции замещения возрастает в следующем ряду галогеналканов: Первичные < Вторичные < Третичные Третичные Галогеналканы вступают в реакцию замещения особенно легко (в отсутствие сильного основания): (СН3)3ССl+Н2O®водный этанол,25°(СН3)3СОН+НСl В целом в присутствии водного раствора основания для первичных галогеналканов характерны в основном реакции отщепления, вторичные Галогеналканы - отщепления и замещения, а третичные образуют главным образом продукты замещения. Галогеналканы вступают в SN-реакции с различными нуклеофилами, такими, как цианид-анион CN-, ацетат-анион СН3СОO-, аммиак:NH3, амины RN..H2 и многие другие. 3. При добавлении раствора галогеналкана в диэтиловом эфире СН3СН2ОСН2СН3 к магниевой стружке происходит экзотермическая реакция: магний переходит в раствор и образуется реактив Гриньяра формулы R—Mg—Г, где R — алкильная или арильная группа, а Г — галоген. RГ+Mg®эфир R-Mg-Г Реактивы Гриньяра вступают в реакции со многими соединениями, что позволяет использовать их в синтезе самых различных веществ. Элиминирование (от лат. elimino — изгоняю) — это отщепление двух атомов или групп атомов от соседних атомов углерода с образованием между ними π-связи. Атомы углерода при элиминировании переходят из sр3- в sp2-гибридное состояние (или из sр2- в sp).Реакция элиминирования может проходит в одну стадию (по механизму E2), либо в две стадии, по механизму E1 Исходными веществами могут служить представители разных классов органических соединений. Зайцева Правило: при дегидратации вторичных и третичных спиртов в присут. сильных к-т и при дегидрогалогенировании вторичных и третичных алкилгалогенидов под действием оснований протон отщепляется преим. от наименее гидрогенизир. атома С с образованием соед. II:Элиминирование по 3. п. протекает в ряде случаев при разложении вторичных и третичных солей диазония (X = N2+), а также вторичных и третичных алкил- и арилсульфонатов (X = AlkSO2O, ArSO2O) под действием оснований. В практике орг. синтеза обычно не встречаются примеры р-ций элиминирования, в к-рых 3. п. выполнялось бы абсолютно; реально получаются смеси изомерных олефинов ф-л II и III. При элиминировании НХ из соед. I получающийся наиб. замещенный олефин (ф-лы II) наз. образующимся по 3. п., а терминальный олефин III – образующимся по правилу Гофмана (см. Гофмана реакции). Элиминированию по 3. п. способствует наличие в соед. I легко удаляющейся группы X; способность к такому отщеплению уменьшается в ряду N2+ > I- > Br- > Cl- > TsO- > R2S+ > F- > R3N+ (Ts – CH3C6H4SO2, R – алифатич. радикал). Напр., при обработке 2-бром-, 2-тозилокси- и 2-диметилсульфониобутана этилатом Na в этаноле соотношения образующихся 2-бутена и 1-бутена составляют соотв. 81:19, 65:35 и 26:74. В р-циях, протекающих под действием алкоголятов, элиминированию по 3. п. благоприятствует применение оснований с радикалом небольшого объема. Так, при использовании С 2 Н 5 ОК, трет-C4H9OK, трет -С 5 Н 11 ОК и (С 2 Н 5)3 СОК в р-ции дегидрогалогенирования 2-метил-2-бромбутана соотношения образующихся соед. IV и V составляют соотв. 62:38, 27:73, 22:78 и 11:89: Увеличение объема радикалов R и R’ в исходном соед. препятствует протеканию р-ции по 3. п., напр.: Образованию олефинов по 3. п. способствует применение биполярных протонных р-рителей. Так, при проведении р-ции в этаноле и ДМСО соотношения продуктов VI и VII составляют соотв. 65:35 и 46:54. Правило сформулировано А. М. Зайцевым в 1875.
 30.Классиф гидроксилпроизводных углеродов, номенклатура.Изомерия. Производные углеводородов, в молекулах которых один или несколько атомов водорода замещены гидроксильными группами (ОН), называют предельными спиртами или алкоголями. Общая формула R-OH. Спирты классифицируются: 1) по строению углеводородного радикала различают: а) спирты алифатического (жирного ряда), Аlk-ОН; б) ароматические, которые разделяются на фенолы Аr-OH и жирноароматические спирты Ar(CH2)n-OH; 2) по числу гидроксилов спирты бывают одно-, двух– и многоатомные. Например: а) одноатомные спирты СН3-ОН (метанол); б) двухатомный спирт HO-CH2-CH2OH (этандиол); в) трехатомный спирт НОСН2-СНОН-СН2ОН (глицерин).В зависимости от характера углеродного атома, при котором находится гидроксил, различают первичные, вторичные и третичные спирты.1) R-CH2-OH, или Аr-СН2-ОН, – первичный спирт;2)– вторичный спирт; 3) – третичный спирт. Изомерия и номенклатура. Изомерия спиртов зависит от строения углеводородной цепи и положения гидроксила в цепи. Спирты часто называют по радикально-спиртовой и систематической (ИЮПАК) номенклатуре. При названии спирта по радикально-спиртовой номенклатуре в основе лежит название соответствующего углеводородного радикала, связанного с гидроксилом, с прибавлением окончания – овый спирт. Например: 1) СН3-ОН – метиловый спирт (древесный); 2) С2Н5-ОН – этиловый спирт; 3) н-С4Н9ОН – бутиловый спирт; 4)
30.Классиф гидроксилпроизводных углеродов, номенклатура.Изомерия. Производные углеводородов, в молекулах которых один или несколько атомов водорода замещены гидроксильными группами (ОН), называют предельными спиртами или алкоголями. Общая формула R-OH. Спирты классифицируются: 1) по строению углеводородного радикала различают: а) спирты алифатического (жирного ряда), Аlk-ОН; б) ароматические, которые разделяются на фенолы Аr-OH и жирноароматические спирты Ar(CH2)n-OH; 2) по числу гидроксилов спирты бывают одно-, двух– и многоатомные. Например: а) одноатомные спирты СН3-ОН (метанол); б) двухатомный спирт HO-CH2-CH2OH (этандиол); в) трехатомный спирт НОСН2-СНОН-СН2ОН (глицерин).В зависимости от характера углеродного атома, при котором находится гидроксил, различают первичные, вторичные и третичные спирты.1) R-CH2-OH, или Аr-СН2-ОН, – первичный спирт;2)– вторичный спирт; 3) – третичный спирт. Изомерия и номенклатура. Изомерия спиртов зависит от строения углеводородной цепи и положения гидроксила в цепи. Спирты часто называют по радикально-спиртовой и систематической (ИЮПАК) номенклатуре. При названии спирта по радикально-спиртовой номенклатуре в основе лежит название соответствующего углеводородного радикала, связанного с гидроксилом, с прибавлением окончания – овый спирт. Например: 1) СН3-ОН – метиловый спирт (древесный); 2) С2Н5-ОН – этиловый спирт; 3) н-С4Н9ОН – бутиловый спирт; 4)
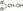
 – трет-бутиловый спирт. В основе названия спирта по ИЮПАК лежит наименование углеводорода самой длинной углеводородной цепи, наличие же гидроксильной группы указывается окончанием – ол, с цифрой за ним, указывающей номер атома углерода, при котором стоит гидроксил. При этом углеродная цепь нумеруются таким образом, чтобы гидроксил имел наименьший номер. Заместительная номенклатура используется достаточно редко. По ней спирты называют как производные карбинола (метанола). Например, фенилкарбинол – бензиловый спирт (оксиметилбензол), этилкарбинол – пропиловый спирт (пропанол), винилкарбинол – пропен-2-ол-1 (аллиловый спирт). Тривиальная номенклатура, напротив, до сих пор широко применяется. Метанол (метиловый спирт, муравьиный спирт), пропиловый спирт, группа амиловых спиртов (С5) и т.д. Не говоря уже о непредельных спиртах, которые практически только и называются тривиальными названиями – аллиловый и пропаргиловый спирты. Изомерия. Изомерия спиртов аналогична изомерии галогенопроизводных. В случае спиртов кроме изменения строения углеродного скелета может изменяться положение –OH группы.
– трет-бутиловый спирт. В основе названия спирта по ИЮПАК лежит наименование углеводорода самой длинной углеводородной цепи, наличие же гидроксильной группы указывается окончанием – ол, с цифрой за ним, указывающей номер атома углерода, при котором стоит гидроксил. При этом углеродная цепь нумеруются таким образом, чтобы гидроксил имел наименьший номер. Заместительная номенклатура используется достаточно редко. По ней спирты называют как производные карбинола (метанола). Например, фенилкарбинол – бензиловый спирт (оксиметилбензол), этилкарбинол – пропиловый спирт (пропанол), винилкарбинол – пропен-2-ол-1 (аллиловый спирт). Тривиальная номенклатура, напротив, до сих пор широко применяется. Метанол (метиловый спирт, муравьиный спирт), пропиловый спирт, группа амиловых спиртов (С5) и т.д. Не говоря уже о непредельных спиртах, которые практически только и называются тривиальными названиями – аллиловый и пропаргиловый спирты. Изомерия. Изомерия спиртов аналогична изомерии галогенопроизводных. В случае спиртов кроме изменения строения углеродного скелета может изменяться положение –OH группы.
31 Способы получ спиртов.Гидролизгалогеналканов. Гидратация алкенов. Наиболее общим способом является получение спиртов из галоидных соединений обменом атомат галоида на гидроксил:  Этим способом можно получать спирты первичные, вторичные и третичные в зависимости от строения радикала СnН2n+1 Иногда для получения спирта из галоидного алкила сначала получают сложный эфир уксусной кислоты взаимодействием галоидного алкила с уксуснокислым серебром, например
Этим способом можно получать спирты первичные, вторичные и третичные в зависимости от строения радикала СnН2n+1 Иногда для получения спирта из галоидного алкила сначала получают сложный эфир уксусной кислоты взаимодействием галоидного алкила с уксуснокислым серебром, например 
а затем омылением сложного эфира получают спирт:

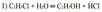 Гидролиза галогеналканов
Гидролиза галогеналканов
Гидратация алкенов: CH2=CH2 + H2O (кат.)  CH3CH2OH Присоединение воды к несимметричным алкенам идет по правилу Марковникова с образованием вторичных и третичных спиртов CH3–CH=CH2 +H2O(кат.) CH3CH(OH)CH3 ---\\\--- (CH3)2C=CH2 + H2O (кат.) (CH3)3C–OH
CH3CH2OH Присоединение воды к несимметричным алкенам идет по правилу Марковникова с образованием вторичных и третичных спиртов CH3–CH=CH2 +H2O(кат.) CH3CH(OH)CH3 ---\\\--- (CH3)2C=CH2 + H2O (кат.) (CH3)3C–OH
Восстановление Альдегиды и кетоны способны восстанавливаться, т. е. присоединять атомы водорода, причем главными продуктами восстановления являются спирты: первичные — из альдегидов и вторичные — из кетонов.Восстановление кетонов часто может идти также и в другом направлении, а именно с присоединением атома водорода к кислородукарбонильной группы, причем атомы углерода карбонильных групп двух молекул связываются между собой, например:
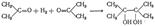
Таким образом получаются двухатомные спирты с удвоенным числом атомов углерода в молекуле, сравнительно с молекулойисходного кетона, — так называемые пинаконы. Возможно и более глубокое восстановление карбонильной группы — замещение атома кислорода двумя водородными атомами(элиминирование карбонильной группы).Взаимодействием реактива Гриньяра с формальдегидом можно получить практически любой первичный спирт (кроме метанола). Для этого продукт присоединения реактива Гриньяра
гидролизуют водой:
Н2O
Н2СО + RMgX → R-CH2-O-MgX → R-CH2-OH.
-Mg(OH)X Оксосинтез является одним из важнейших нефтехимических процессов, с помощью которого производится широкий ассортимент кислородсодержащих соединений - альдегидов, спиртов, кислот, сложных эфиров, кетонов и др. Эти продукты могут быть получены на основе доступных дешевых источников сырья - газов крекинга и пиролиза, продуктов дегидрирования парафинов, жидких продуктов термической переработки нефти, полимер-олефинов.Реакция гидроформилирования основана на взаимодействий олефинов с окисью углерода и водородом. Катализаторами реакции являются карбонилы кобальта, а также карбонилы других металлов VIII группы таблицы Менделеева.Наиболее признанный механизм: 
32Хим св-ва спиртов.Р. с сохран. Атома кислорода. Алкоголята. Спирты - органические соединения, содержащие одну или более гидроксильных групп(гидроксил, −OH), непосредственно связанных с насыщенным (находящемся в состоянии sp³ гибридизации) атомом углерода[1]. Спирты можно рассматривать как производные воды (H−O−H), в которых один атом водорода замещен на органическуюфункциональную группу: R−O−H. В номенклатуре IUPAC для соединений, в которых гидроксильная группа связана с ненасыщенным находящемся в состоянии sp2гибридизации атомом углерода, рекомендуются названия «енолы» (гидроксил связан с винильной C=C связью)[2] и «фенолы» (гидроксил связан с бензольным или другим ароматическим циклом). Реакция альдегидов со спиртами. Синтез полуацеталей и ацеталей. В благоприятных условиях (например: а) при нагревании с кислотой или в присутствии водоотнимающих средств; б) при внутримолекулярной конденсации с образованием пяти- и шестичленных циклов) альдегиды реагируют со спиртами. При этом к одной молекуле альдегида может присоединиться либо одна молекула спирта (продукт – полуацеталь), либо две молекулы спирта (продукт – ацеталь): 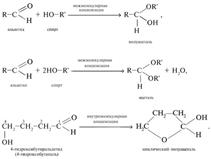
Кисло́ты — сложные вещества, в состав которых обычно входят атомы водорода, способные замещаться на атомы металлов, икислотный остаток. Водные растворы кислот имеют кислый вкус, обладают раздражающим действием, способны менять окраскуиндикаторов, отличаются рядом общих химических свойств. АЛКОГОЛЯТЫ, продукты замещения атома Н в молекуле спирта на металл (М). Алкоголятыодноатомных спиртов. Их общая ф-ла M(OR)n, где n-степень окисления металла. Алкоголяты щелочных, щел.-зем.металлов, Т1(1) и первичных спиртов-ионные соед.; неплавки, нелетучи; т. разл. 200-300 °С; раств. в спиртах и жидком NH3;электролиты в р-ре. Из спиртовых р-ров обычно выделяются в виде кристаллосольватов. Производные металлов III-VIII групп и спиртов(начиная с С2Н5ОН), а также M1OR-mpem- молекулярные мономерные или олигомерные соед.; имеют низкие т-ры плавления и кипения; хорошо раств. в неполярных р-рителях, плохо-в спиртах; р-ры не проводят ток. Метилаты тех же элементов-обычно координац.полимеры; неплавки, нелетучи; не раств. ни в одном из р-рителей. Большинство алкоголятов элементов середины периодич. системы сочетают св-ва ионных и молекулярных соед. Все алкоголяты очень гигроскопичны. Образование алкоголятов Водородный атом гидроксильной группы при действии щелочных металлов способен заменяться наатомы этих металлов, причем получаются твердые, растворимые в спирте соединения, называемые алкоголятами:
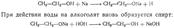
Первичные спирты при окислении образуют альдегиды, которые затем легко окисляются до карбоновых кислот.
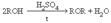
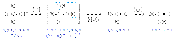
При окислении вторичных спиртов образуются кетоны.
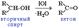
Третичные спирты более устойчивы к действию окислителей. Они окисляются только в жестких условиях (кислая среда, повышенная температура), что приводит к разрушению углеродного скелета молекулы и образованию смеси продуктов (карбоновых кислот и кетонов с меньшей молекулярной массой).Процесс идет через стадию дегидратации спирта с последующим деструктивным (жестким) окислением алкена. Например:
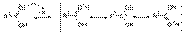 33Хим св-ва спиртов. Нуклеофил замещ гидроксо группы. Взаимодействие спиртов с галогенводородными кислотами. Спирты - органические соединения, содержащие одну или более гидроксильных групп(гидроксил, −OH), непосредственно связанных с насыщенным (находящемся в состоянии sp³ гибридизации) атомом углерода[1]. Спирты можно рассматривать как производные воды (H−O−H), в которых один атом водорода замещен на органическуюфункциональную группу: R−O−H. В номенклатуре IUPAC для соединений, в которых гидроксильная группа связана с ненасыщенным находящемся в состоянии sp2гибридизации атомом углерода, рекомендуются названия «енолы» (гидроксил связан с винильной C=C связью)[2] и «фенолы» (гидроксил связан с бензольным или другим ароматическим циклом) Реакции нуклеофильного замещения ОН группы Для проведения SN-реакций в спиртах необходимо предварительно модифицировать ОН группу, превратив ее в хорошую уходящую группу. Для этого спирты переводят в оксониевые соединения или эфиры неорганических кислот:
33Хим св-ва спиртов. Нуклеофил замещ гидроксо группы. Взаимодействие спиртов с галогенводородными кислотами. Спирты - органические соединения, содержащие одну или более гидроксильных групп(гидроксил, −OH), непосредственно связанных с насыщенным (находящемся в состоянии sp³ гибридизации) атомом углерода[1]. Спирты можно рассматривать как производные воды (H−O−H), в которых один атом водорода замещен на органическуюфункциональную группу: R−O−H. В номенклатуре IUPAC для соединений, в которых гидроксильная группа связана с ненасыщенным находящемся в состоянии sp2гибридизации атомом углерода, рекомендуются названия «енолы» (гидроксил связан с винильной C=C связью)[2] и «фенолы» (гидроксил связан с бензольным или другим ароматическим циклом) Реакции нуклеофильного замещения ОН группы Для проведения SN-реакций в спиртах необходимо предварительно модифицировать ОН группу, превратив ее в хорошую уходящую группу. Для этого спирты переводят в оксониевые соединения или эфиры неорганических кислот:
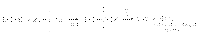
В биохимических процессах замещение ОН группы протекает через стадию превращения спиртов в эфиры фосфорной, дифосфорной и трифосфорной кислот, анионы которых являются чрезвычайно легко уходящими группами: Замещение ОН группы на галоген Спирты взаимодействуют с галогеноводородами и галогенидами фосфора и серы с образованием галогенпроизводных. 1. Действие галогеноводородов. R-OH + HHal ® R-Hal + H2O Реакция протекает через стадию протонирования ОН группы с последующим нуклеофильным замещением по механизму SN1 илиSN2. 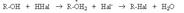
Первичные спирты реагируют по механизму SN2. Реакция идет медленно, так как в протонных растворителях (вода, спирты) галогенид-ионы – слабые нуклеофилы. Вторичные и третичные спирты реагируют по механизму SN1. Таким образом, реакционная способность спиртов возрастает в ряду: первичный, вторичные, третичные Реакционная способность галогеноводородов увеличивается в ряду: HCl параллельно с возрастанием силы кислот и нуклеофильности галогенид-анионов. Однако HI не может быть использована, так как легко восстанавливает спирты до углеводородов. HBr реагирует с первичными, вторичными и третичными спиртами. HCl действует только на третичные спирты.
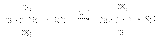
Для вторичных и первичных спиртов требуется присутствие катализатора – кислоты Льюиса.
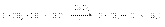
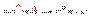
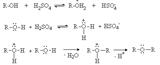
 34Хим св-ва спиртов. Взаимодействие спиртов с серной кислотой: Образование простых эфиров При нагревании спиртов в присутствии серной кислоты происходит замещение ОН группы на группу OR и образуются простые эфиры
34Хим св-ва спиртов. Взаимодействие спиртов с серной кислотой: Образование простых эфиров При нагревании спиртов в присутствии серной кислоты происходит замещение ОН группы на группу OR и образуются простые эфиры
Реакция протекает через стадию протонирования спирта с последующим нуклеофильным замещением, в котором одна молекула спирта (протонированная) выполняет роль субстрата, а вторая – нуклеофила. В отличие от спиртов, являющихся слабыми нуклеофилами, алкоголяты, образующие алкоксид-ионы RO− — сильные нуклеофилы и легко реагируют с алкилгалогенидами по механизму SN 2 с образованием простых эфиров:Вместо алкилгалогенидов можно использовать также алкилсульфонаты Побочными продуктами реакции являются алкены, образующиеся в результате конкурирующего процесса элиминирования спирта:
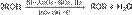
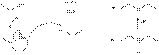
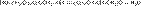 Межмолекулярная и внутримолекулярная дегидратация спиртов При осторожном нагревании в присутствии серной кислоты происходит межмолекулярная дегидратация спиртов с образованием простых эфиров. Если в реакцию с кислотой вступают двухатомные спирты, будет протекать реакция внутримолекулярной дегидратации с образованием гетероциклических соединений. Так например, 1,4-бутандиол образует тетрагидрофуран Так как реакция получения эфира обратима, для её смещения вправо обычно используют метод отгонки конечных продуктов (воды или эфира) из реакционной смеси. Существуют и методы термокаталитической дегидратации спиртов. Например, первичные спирты в присутствии смешанного Ni−Al2O3−SiO2 катализатора и водорода при нагревании превращаются в простые эфиры:
Межмолекулярная и внутримолекулярная дегидратация спиртов При осторожном нагревании в присутствии серной кислоты происходит межмолекулярная дегидратация спиртов с образованием простых эфиров. Если в реакцию с кислотой вступают двухатомные спирты, будет протекать реакция внутримолекулярной дегидратации с образованием гетероциклических соединений. Так например, 1,4-бутандиол образует тетрагидрофуран Так как реакция получения эфира обратима, для её смещения вправо обычно используют метод отгонки конечных продуктов (воды или эфира) из реакционной смеси. Существуют и методы термокаталитической дегидратации спиртов. Например, первичные спирты в присутствии смешанного Ni−Al2O3−SiO2 катализатора и водорода при нагревании превращаются в простые эфиры:
Метод межмолекулярной дегидратации — один из наиболее старых способов получения эфиров — используется весьма ограниченно и только для неразветвлённых первичных спиртов из-за высокой доли алкенов, образующихся в случае внутримолекулярной дегид5ратации при использовании вторичных и третичных спиртов. Спирты способны образовывать сложные эфиры в реакциях с органическими кислотами при нагревании в присутствии кислотного катализатора (как правило, концентрированной H2SO4). Этот процесс получил название кислотно-каталитической реакции этерификации (также известен как реакция Фишера). Например, взаимодействие этанола с уксусной кислотой дает этилацетат:
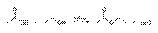 Механизм реакции:
Механизм реакции:
 Кислотно-каталитическая реакция этерификации — простейший и наиболее удобный метод получения сложных эфиров для случая, когда ни кислота, ни спирт не содержат чувствительных функциональных групп. В качестве катализатора, помимо традиционно используемой серной кислоты, могут выступать кислота Льюиса или Бренстеда; растворителем, обычно, служит сам спирт или, если это невозможно — толуол или ксилол. Для увеличения выхода эфира используют отгонку или химическое связывание воды, а также специализированное лабораторное оборудование — аппарат Дина — Старка. Реакция между спиртом и кислотой происходитвприсутствии дициклогексилкарбодиимида (ДЦК)и небольших количеств 4-N,N-диметиламинопирнидина. ДЦК и карбоновая кислота на первом этапе образует O-ацилизомочевинный интермедиат, который в дальнейшем вступает в реакцию со спиртом, образуя сложный эфир:
Кислотно-каталитическая реакция этерификации — простейший и наиболее удобный метод получения сложных эфиров для случая, когда ни кислота, ни спирт не содержат чувствительных функциональных групп. В качестве катализатора, помимо традиционно используемой серной кислоты, могут выступать кислота Льюиса или Бренстеда; растворителем, обычно, служит сам спирт или, если это невозможно — толуол или ксилол. Для увеличения выхода эфира используют отгонку или химическое связывание воды, а также специализированное лабораторное оборудование — аппарат Дина — Старка. Реакция между спиртом и кислотой происходитвприсутствии дициклогексилкарбодиимида (ДЦК)и небольших количеств 4-N,N-диметиламинопирнидина. ДЦК и карбоновая кислота на первом этапе образует O-ацилизомочевинный интермедиат, который в дальнейшем вступает в реакцию со спиртом, образуя сложный эфир: 

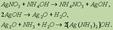 35.Функц. карбонильные группы. Классиф. карбон. соединений.Гомолог. ряды альдегидов и кетонов. Ном-тура и изомерия. Р-ции окисл. алкенов; ок-ия и дегидрирования алканолов; гидролиз дигалогеналкан гидратация алкинов; оксосинтез; пиролиз кальциевых солей карбоновых кислот, пинаколиновая перегруппировка.Карбонильные соединения содержат в молекуле карбонильную группу Карбонильные соединения делятся на альдегиды и кетоны. Свойства альдегидов и кетонов определяются строением карбонильной группы C=O.Систематические названия альдегидов строят по названию соответствующего углеводорода и добавлением суффикса -аль. Нумерацию цепи начинают с карбонильного атома углерода. Тривиальные названия производят от тривиальных названий тех кислот, в которые альдегиды превращаются при окислении. Названия альдегидов по заместительной номенклатуре в соответствии с правилами ИЮПАК производят из названия соответствующего углеводорода с добавлением окончания -аль. Перед корнем названия записывают боковые заместители с указанием их положения их числа. Нумерация атомов углерода начинается с углеродного атома карбонильной группы.Получение: 1. Гидратация алкинов. Hg2+, H+ ---\\\--- СH≡СН + Н2О → СН3-СН=О.2. Общий способ получения карбонильных соединений окисление спиртов. СН3-СН2-ОН + СuО t→ СН3-СН=О + Сu + Н2О.3. При щелочном гидролизе дигалогеналканов, содержащих два атома галогена при одном атоме углерода, образуются двухатомные спирты, в которых две группы ОН соединены с одним атомом углерода. Эти вещества неустойчивы и отщепляют воду, превращаясь в карбонильные соединения:СН3-СНСl2→[СН3СН(ОН)2] 2NaOH СН3-СН=О +Н2О. 4. Дегидрирование спиртов. ZnO, 380° С . СН3-СН(ОН)-СН3 → СН3-СО-СН3
35.Функц. карбонильные группы. Классиф. карбон. соединений.Гомолог. ряды альдегидов и кетонов. Ном-тура и изомерия. Р-ции окисл. алкенов; ок-ия и дегидрирования алканолов; гидролиз дигалогеналкан гидратация алкинов; оксосинтез; пиролиз кальциевых солей карбоновых кислот, пинаколиновая перегруппировка.Карбонильные соединения содержат в молекуле карбонильную группу Карбонильные соединения делятся на альдегиды и кетоны. Свойства альдегидов и кетонов определяются строением карбонильной группы C=O.Систематические названия альдегидов строят по названию соответствующего углеводорода и добавлением суффикса -аль. Нумерацию цепи начинают с карбонильного атома углерода. Тривиальные названия производят от тривиальных названий тех кислот, в которые альдегиды превращаются при окислении. Названия альдегидов по заместительной номенклатуре в соответствии с правилами ИЮПАК производят из названия соответствующего углеводорода с добавлением окончания -аль. Перед корнем названия записывают боковые заместители с указанием их положения их числа. Нумерация атомов углерода начинается с углеродного атома карбонильной группы.Получение: 1. Гидратация алкинов. Hg2+, H+ ---\\\--- СH≡СН + Н2О → СН3-СН=О.2. Общий способ получения карбонильных соединений окисление спиртов. СН3-СН2-ОН + СuО t→ СН3-СН=О + Сu + Н2О.3. При щелочном гидролизе дигалогеналканов, содержащих два атома галогена при одном атоме углерода, образуются двухатомные спирты, в которых две группы ОН соединены с одним атомом углерода. Эти вещества неустойчивы и отщепляют воду, превращаясь в карбонильные соединения:СН3-СНСl2→[СН3СН(ОН)2] 2NaOH СН3-СН=О +Н2О. 4. Дегидрирование спиртов. ZnO, 380° С . СН3-СН(ОН)-СН3 → СН3-СО-СН3
5. Окисление алкенов. Альдегиды и кетоны получают окислением углеводородов ряда этилена кислородом воздуха в присутствии хлоридов палладия (II) и меди (I), например: СH2-CH2+1/2O2 PBCL2*CUCL2 CH3CH=O Этим экономичным способом в промышленности получают низшие альдегиды и кетоны. 6. (оксосинтез), присоединение оксида углерода и водорода к олефинам в присутствии катализатора с образованием альдегидов, например СН3СН = СН2 + СО + Н2→СН3СН2СН2СНО. 7/ Пинаколиновая перегруппировка, образ-ие кетонов при действии кислот (HCI, H2SO4), а также ZnCl2 на пинаконы; при этом происходит дегидратация, сопровождающаяся изменением скелета молекулы — миграцией одного из заместителей к соседнему углеродному атому. Отход гидроксильной группы и перемещение заместителя происходят синхронно 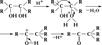
36.Хим св альдегидов и кетонов. Реакции нуклеофильного присоединения. Альдегиды и кетоны, обладая электрофильным центром, способны вступать во взаимодействие с нуклеофильными реагентами. Для оксосоединений наиболее характерны реакции, протекающие по механизму нуклеофильного присоединения обозначаемому АN. Реакция с циановодорооной (синильной) кислотой. приводящее к образованию a- оксинитрилов.Эта реакция используется для удлинения углеродной цепи и получения a- оксикислот. СH3-СH=O+H-CN CH3-CH(CN)-OH Присоединение гидросульфитов служит для выделения альдегидов из смесей с другими веществами и для получения их в чистом виде, поскольку полученное сульфопроизводное очень легко гидролизуется:
R-CH=O+NaHSO3 R-CH-SO3Na
|
OH
Присоединение металлоорганических соединений При этой реакции углеводородный радикал металлоорганического соединения присоединяется к углероду, а остальная часть его молекулы — к кислороду, например:
CH3-CH=O+R-MGI CH3-CH-OMg
|
R
При разложении полученных соединений водой получаются соответствующие спирты.
Наряду с этой реакцией, при действии магнийорганических соединений на кетоны происходит изомеризация кетонов в ненасыщенные спирты и образование смешанных магниевых алкоголятов этих спиртов (Гриньяр):
37.Хим. св-ва альдегид. и кетонов. О-В реакции. Восстановление альдегидов и кетонов в спирты. При восстановлении альдегидов образуются первичные, а при восстановлении кетонов — вторичные спирты
O
// 2H
H3C—C H3C—CH2OH
\ H
уксусный этиловый альдегид спирт
2H
H3C—CO—CH3 H3C—CH—CH3
|
OH
ацетон изопропиловый
спирт
Одной из качественных реакций для обнаружения альдегидной группы является реакция “серебряного зеркала” — окисление альдегидов оксидом серебра.
В растворе аммиака оксид серебра образует комплексное соединение, при действии которого на альдегид происходит окислительно-восстановительная реакция. Альдегид окисляется в соответствующую кислоту (точнее, в ее аммонийную соль), а комплексный катион восстанавливается до металлического серебра, которое дает блестящий налет на стенках пробирки — “серебряное зеркало”:
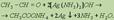
Другая качественная реакция на альдегиды заключается в окислении их гидроксидом меди (II). При окислении альдегида светло-голубой гидроксид меди (II) превращается в желтый гидроксид меди (I) при комнатной температуре. Если подогреть раствор, то гидроксид меди (I) превращается в оксид меди (I) красного цвета, который плохо растворим в воде и выпадает в осадок:

При прибавлении фуксинсернистой кислоты к раствору альдегида смесь приобретает красное или красно-фиолетовое окрашивание. При последующем прибавлении минеральных кислот это окрашивание, как правило, исчезает; исключение составляет формальдегид; окрашивание фуксинсернистой кислоты, вызванное формальдегидом, не исчезает от прибавления кислот.
38.Хим св-ва альдегидов и кетонов. Конденсация. Хим св-ва обусловлены наличием в их молекулах сильно полярной карбонильной группы (связь 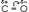 поляризована в сторону атома кислорода). Чем больше частичный заряд(
поляризована в сторону атома кислорода). Чем больше частичный заряд(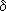 +)на атоме C этой группы, тем выше активность соединения.1.Горение: 2CH3CHO+5O2=4CO2+4Н2О 2CH3COCH3 + 9O2
+)на атоме C этой группы, тем выше активность соединения.1.Горение: 2CH3CHO+5O2=4CO2+4Н2О 2CH3COCH3 + 9O2 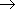 6CO2 + 6H2O 2.Присоединение(по двойной связи карбонилгруппы). В ряду HCHO RCHO RCOR' склонность к реакциям присоединения уменьшается. Это связано с наличием и числом углеводородных радикалов, связанных с атомом С карбонильной группы. а) Гидрирование (восстановление водородом): HCHO + H2
6CO2 + 6H2O 2.Присоединение(по двойной связи карбонилгруппы). В ряду HCHO RCHO RCOR' склонность к реакциям присоединения уменьшается. Это связано с наличием и числом углеводородных радикалов, связанных с атомом С карбонильной группы. а) Гидрирование (восстановление водородом): HCHO + H2 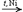 CH3OH
CH3OH
CH3—CO—CH3 + H2 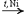 CH3—CH(OH)—CH3 Из альдегидов при этом получаются первичные спирты, а из кетонов - вторичные. 3)Окисление: CH3CHO + Ag2O
CH3—CH(OH)—CH3 Из альдегидов при этом получаются первичные спирты, а из кетонов - вторичные. 3)Окисление: CH3CHO + Ag2O 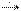 2Ag
2Ag  + CH3COOH (р-ция "серебряного зеркала" – качеств реакция)
+ CH3COOH (р-ция "серебряного зеркала" – качеств реакция)
HCHO + 2Cu(OH)2 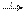 2H2O + Cu2O
2H2O + Cu2O 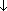 + HCOOH (образуется красный осадок - качественная реакция).Кетоны слабыми окислителями не окисляются.4.Замещение атомов водорода в углеводородном радикале (замещение происходит в
+ HCOOH (образуется красный осадок - качественная реакция).Кетоны слабыми окислителями не окисляются.4.Замещение атомов водорода в углеводородном радикале (замещение происходит в  -положение, т. е. замещается атом водорода у 2-го атома углерода):
-положение, т. е. замещается атом водорода у 2-го атома углерода):
| 2(a) | |||
| CH3 | —CH2 | —CHO | + Cl2  CH3—CHCl—CHO + HCl CH3—CHCl—CHO + HCl |
АЛЬДОЛЬНАЯ КОНДЕНСАЦИЯ, взаимод. Двух молекул альдегида или кетона (одинаковых или разных) в присут. к-т или оснований с образованием 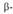 гидроксиальдегидов (альдолей), напр.:
гидроксиальдегидов (альдолей), напр.:
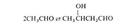 Р-ция обратима и может осуществляться только при наличии хотя бы у одного реагента атома Н в
Р-ция обратима и может осуществляться только при наличии хотя бы у одного реагента атома Н в 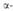 положении к карбонильной группе.Кетоны реагируют значительно труднее альдегидов.
положении к карбонильной группе.Кетоны реагируют значительно труднее альдегидов.
КРОТOНОВАЯ КОНДЕНСАЦИЯ, взаимодействие двух молекул альдегидов или кетонов друг с другом в присут. оснований или к-т, сопровожд отщеплением воды и образованием -непредельного карбонильного соед.Первая стадия процесса - синтез альдоля, к-рый обычно при комнатной т-ре или при нагр. дегидратируется. Кетоны реагируют в более жестких условиях, чем альдегиды. Образующееся в р-ции нeнасыщ. карбонильное соед. может вступать в р-цию с исходными в-вами, напр
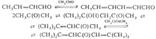
Механизм дегидратации альдоля зависит от природы катализатора (основания или к-ты), напр.:
 Кротоновую конденсацию широко используют для синтеза ненасыщ. альдегидов и кетонов (напр., для пром. получения кротонового альдегида).
Кротоновую конденсацию широко используют для синтеза ненасыщ. альдегидов и кетонов (напр., для пром. получения кротонового альдегида).
39.Амины. Классиф, номенкл, изомер. Амины-производные аммиака, в молекуле которого 1,2 или 3 атома водорода замещены органическими радикалами. Радикалы могут быть алифатическими (насыщенными или ненасыщ), карбоциклическими, ароматическими или гетероциклическими. В зависимости от числа органических радикалов у атома азота амины делятся на первичные - RNH2, вторичные - R2NH и третичные – R3N. При этом не имеет значения, какие органические радикалы (первичные, вторичные или третичные) выступают в роли заместителя – в первичных, вторичных и третичных аминах могут присутствовать как первичные, так и вторичные и третичные алкильные радикалы. Существуют также четвертичные соли аммония R4N+X- - производные иона аммония, у которого все валентности атома азота заняты органическими заместителями.Для обозначения аминов используют 3вида номенклатуры – тривиальную (напр, анилин, толуидин), заместительную (метиламин, триметиламин) и номенклатуру ИЮПАК (1-аминогексан). Для обозначения простейших аминов наиб часто применяют заместительную номенклатуру, согласно которой названия аминов строятся путем перечисления углеводородных радикалов в алфавитном порядке с добавлением окончания амин. В более сложных случаях используют номенклатуру ИЮПАК, в которой амины рассматривают как производные углеводородов с префиксом амино-. В классе аминов существуют след виды изомерии: изомерия углеводородного скелета (напр, 1-аминобутан и 1-амино-2-метилпропан), положения аминогруппы (напр, 1- и 2-аминобутаны), для полизамещенных ароматических аминов – изомерия взаимного расположения заместителей (напр, орто-, мета- и пара-толуидины) и изомерия первичных, вторичных и третичных аминов (напр, н-пропиламин, метилэтиламин и триметиламин).1- Наиболее общим методом получения первичных аминов является восстановление нитросоединений: 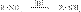 . Важнейший ароматический амин - анилин - образуется при восстановлении нитробензола (восстановители - водород в присутствии металлических катализаторов, Fe + HCl, сульфиды):
. Важнейший ароматический амин - анилин - образуется при восстановлении нитробензола (восстановители - водород в присутствии металлических катализаторов, Fe + HCl, сульфиды): 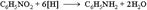 Эта реакция носит имя русского химика Н.Н. Зинина, осуществившего ее впервые в 1842 г. 1) Восстановление амидов (восстановитель - алюмогидрид лития LiAH4):
Эта реакция носит имя русского химика Н.Н. Зинина, осуществившего ее впервые в 1842 г. 1) Восстановление амидов (восстановитель - алюмогидрид лития LiAH4): 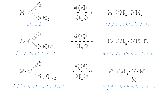 Восстановление нитрилов с образованием первичных аминов:R-C
Восстановление нитрилов с образованием первичных аминов:R-C 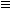 N + 4[H] R-CH2NH2 Этим способом в промышленности получают гексаметилендиамин, который используется в производстве полиамидного волокна найлон.
N + 4[H] R-CH2NH2 Этим способом в промышленности получают гексаметилендиамин, который используется в производстве полиамидного волокна найлон.  Получение аминов путём вве
Получение аминов путём вве