2015-06-10
2015-06-10 525
525Если дыхание в норме и нет существенных отклонений от нормы пульса и артериального давления, после того как получены достаточно исчерпывающие данные анамнестического, характера и в основном решены проблемы элементарной терапии, необходимо провести быстрое, но осторожное дифференцированное обследование больного. Целью обследования должно быть получение данных, не только объясняющих происхождение комы, но и обусловливающих дальнейший ход лечения и, поддержания витальных функций организма.
Выяснению природы комы, возможной локализации поражения мозга может способствовать оценка уровня сознания, характера дыхания, данных исследования состояния глазных яблок и зрачков, характера мышечного ответа на неприятные (болевые) раздражения.
Рассмотрим более подробно каждый из этих признаков.
Уровень сознания. У больных, не находящихся в глубоком коматозном состоянии, уровень сознания является наиболее чувствительным показателем тенденций в развитии комы, т. е. показателем предстоящего улучшения или ухудшения. Для оценки сознания удобнее всего пользоваться шкалой комы Глазго. Больного необходимо обследовать с использованием этой шкалы каждые 2 ч. Уменьшение числа баллов, получаемых при оценке сознания по этой шкале, свидетельствует об ухудшении тенденций в развитии комы. Наш опыт показывает, что изменения уровня сознания и их направленность являются более тонким и достоверным показателем комы, чем, например, характер дыхательной функции и даже ЭЭГ.
Расстройства дыхания. Нарушения деятельности мозга, в частности ствола мозга, вызывают расстройства дыхания, которые могут быть как самостоятельной причиной, так и вспомогательным фактором в развитии сопора или комы.
Классическим примером подобных расстройств является дыхание Чейна—Стокса. В некоторых случаях это первое дыхательное нарушение, свидетельствующее о депрессии мозга. Существует много причин возникновения дыхания типа Чейна—Стокса: атеросклероз мозговых сосудов, нарушение кровообращения мозга при застойной недостаточности и др.
Центральная нейрогенная гипервентиляция, свидетельствующая, как правило, о поражении среднего мозга, обычно характеризуется устойчивым, быстрым, глубоким и регулярным дыханием. Иногда такое дыхание называют машинным. В чистой форме, т. е. без гипоксии или гиперкапнии, нарушения дыхания наблюдаются редко.
Наш многолетний опыт лечения коматозных состояний и консультаций больных, находящихся в коме в связи с каким-либо критическим состоянием, показывает, что лечащие врачи часто не распознают элементарных расстройств механики дыхания у больного, принимая патологически экспрессивный характер дыхательных движений за норму. Чаще всего «просматривают» так называемое парадоксальное дыхание, когда у больного нарушается синхронность реберного и диафрагмального компонентов дыхания или полностью выпадает диафрагмальный компонент. Именно в подобных ситуациях у больного возникает форсированное реберное дыхание, создающее для малоосведомленного наблюдателя иллюзию высокой дыхательной активности и, следовательно, адекватности дыхательной функции в целом. Тонкие неврологические механизмы возникновения описанного варианта дыхательных расстройств, характеризующихся торможением диафрагмального звена, изучены недостаточно. Ясно, однако, что они являются следствием биохимического или анатомического поражения структур среднего мозга. Развитие при этом форсированной реберной дыхательной активности следует рассматривать как выражение компенсации функции. Подобный вариант нарушения дыхания не всегда является показанием к ИВЛ. Такая необходимость возникает лишь при сочетании его с выраженной комой. Наиболее часто торможение диафрагмальной активности наблюдается как рефлекторное явление при перитоните у больного в ясном сознании и в этих случаях не требует ИВЛ. Опытный врач обязан различать эти варианты дыхательных расстройств.
Апнеистический вариант дыхания характеризуется паузами в конце инспираторной фазы. Такое дыхание нетипично для комы, но если наблюдается, то свидетельствует о возможном поражении в зоне моста мозга.
Атаксический вариант дыхания характеризуется нерегулярными альтернациями по частоте и по глубине. Обычно он указывает на депрессию ствола мозга и, как правило, требует искусственного поддержания дыхательной функции, т. е. ИВЛ.
Подобные нарушения дыхательной функции, являющиеся следствием первичного поражения зон мозга, редко возникают в описанной классической форме. Однако они, как правило, развиваются у больных с поражением мозга при выраженном набухании его или при глубоких метаболических энцефалопатиях и предшествуют полному прекращению дыхания.
Несколько другая картина нарушения дыхательной активности наблюдается при передозировке седативных веществ. Диффузная депрессия ствола проявляется постепенным снижением частоты дыхания и его глубины без каких-либо других отклонений. Глубокое медикаментозное угнетение дыхания, как правило, ведет к развитию гипоксии мозга.
Справедливость требует подчеркнуть, что описанные варианты расстройств дыхательной функции в клинической практике наблюдаются не столь часто. Это связано с тем, что показания к ИВЛ, требующей, как правило, выключения спонтанного дыхания, определяются прежде всего наличием гипоксии (гипоксемии), а не расстройствами механики дыхания. Чаще всего к моменту возникновения дыхательных расстройств гипоксия достаточно выражена и ИВЛ уже начата.
Исследование глаз — очень важный элемент в оценке комы. Как уже отмечалось, одинаковые по величине, нормально реагирующие зрачки свидетельствуют о том, что кома не имеет связи с деструкцией мозга и обусловлена скорее всего метаболическими или токсическими причинами. Если зрачки одинаковых размеров, но не реагируют на свет, то больше оснований предполагать структурные или глубокие токсико-метаболические причины комы.
Выраженная неравномерность зрачков, особенно если она сочетается с различной реакцией на свет, является прямым указанием на одностороннее поражение мозга.
Расширение и фиксация зрачка на одном глазу могут указывать на вероятность ущемления мозга в тенториальном отверстии на стороне большего зрачка. Такое ущемление приводит к сдавлению III черепного нерва.
Для представления о функциональной интегрированности ствола мозга важно оценить рефлекторные сопряженные движения глазных яблок. Полное их отсутствие указывает на глубокую кому и реальную возможность прекращения дыхания или по крайней мере на его возможную депрессию. В случае глубокого отравления барбитуратами (или другими седативными, препаратами) глазные рефлексы при калорической пробе могут отсутствовать. Однако это не исключает возможности пробуждения больного через сутки или позже. Отсутствие глазодвигательных рефлексов при немедикаментозной коме — очень неблагоприятный прогностический признак.
Реакции скелетных мышц можно оценивать по характеру мышечных ответов на болевое раздражение, например на давление в области надбровных дуг, угла нижней челюсти, на грудину или постукивание молоточком между пальцами. Ответы могут быть различными — от адекватной двигательной или речевой реакции на раздражение до полного отсутствия реакций. Промежуточными вариантами могут быть гримасы и попытки движений (сгибание рук, разгибание ног), признаки: децеребрации в виде ротационных движений конечностей и др.
В части случаев при коме не возникает необходимости в поясничной пункции. Однако она должна быть выполнена при; тяжелой черепно-мозговой травме, геморрагическом инсульте, у инфекционных больных, а также в случае комы, развивающейся на фоне солитарных или распространенных нагноительных процессов (абсцесс, эмпиема плевры и т. д.). Иногда бывает целесообразно пунктировать субдуральное пространство, чтобы определить степень развития отека мозга.
Диагностика отека мозга. У больных, находящихся в крайне тяжелых (критических) состояниях, кома, как правило, развивается постепенно и определение ее конкретной причины редко вызывает затруднения. Наиболее часто коматозные состояния в подобных случаях обусловлены ишемией и гипоксическим поражением мозга, интоксикацией, расстройством водно-электролитного баланса организма, нарушением осмоляльности и КОД в сосудистом, интерстициальном пространствах, внутри клеток. Нередко эти причины сочетаются и четко разграничить их невозможно.
Различные варианты поражения мозга — травматического, метаболического, инфекционного, опухолевого, постишемического, непосредственно гипоксического характера часто сопровождаются развитием отека мозга. В связи с этим основной терапевтической проблемой становится ликвидация самого отека.
Различают два варианта отека мозга при коме — цитотоксический и вазогенный. Иногда эти варианты сочетаются или переходят один в другой [Thurel С. et al., 1983].
При цитотоксическом отеке мозга основной механизм связан с расстройством клеточного метаболизма и нарушением транспорта ионов через клеточные мембраны. Первоначально процесс выражается в потере клеткой электролитов, главным образом К+, и замене его Na+ из внеклеточного пространства. При гипоксических состояниях пировиноградная кислота восстанавливается до молочной [Laborit H., 1970] и нарушается деятельность редокс-системы, ответственной за выведение Na+ из клетки,— возникает блокада так называемого натриевого насоса. Мозговая клетка, содержащая повышенное количество Na+, начинает усиленно накапливать воду — появляется отек мозга. Содержание лактата свыше 6—8 ммоль/л в оттекающей от мозга крови всегда свидетельствует об его отеке. Цитотоксическая форма отека мозга всегда генерализована. Она распространяется практически на все отделы, включая стволовые, поэтому довольно быстро начинает проявляться нарушением витальных функций.
Основой вазогенного отека мозга являются, как правило, поражение сосудистой стенки и нарушение гематоэнцефалического барьера. Имеется множество факторов, приводящих к повреждению сосудистой стенки,— прежде всего инсульт, а также травма мозга, опухоль его и др. В результате поражения стенки сосудов плазма крови вместе с содержащимися в ней электролитами и белками покидает сосудистое пространство и пропитывает периваскулярные зоны мозга.
Таким образом, цитотоксический отек — это преимущественно набухание клеточных структур мозга, а вазогенный — мозаичное, неравномерно выраженное накопление плазмоподобной жидкости в интерстициальном пространстве мозга. Однако различия вариантов отека мозга имеют лишь теоретический интерес, так как в большинстве случаев трудно отличить один вариант от другого [Арутюнов А.И., 1960].
При гипоксии мозга любого происхождения, а также при элементарной ишемии его преобладает, естественно, цитотоксический вариант отека мозга. Процесс характеризуется набуханием клеток мозга с последующим повреждением их внутренних структур, в том числе митохондрий [Fujimoto Т. et al., 1976; Hossmann K., 1976].
В первой фазе развития процесс происходит при неповрежденном гематоэнцефалическом барьере и связан прежде всего с расстройством деятельности клеточной натриевой помпы: накопление в клетке L— и аминокислот и повышение осмоляльности способствуют задержке воды в ней — возникает набухание клетки. Во второй фазе постгипоксического, а также ишемического повреждения развивается сосудистый компонент, нарушается гематоэнцефалический барьер. В результате часть плазменных белков покидает сосудистое русло и перемещается в интерстициальное пространство, увлекая за собой воду.
Концентрация лактата в оттекающей от мозга крови может в определенной степени быть дифференциально-диагностическим показателем при цитотоксическом и вазогенном вариантах отека мозга. При метаболическом повреждении мозга и, следовательно, истинном набухании клеток содержание лактата в крови всегда высокое.
В диагностике отека мозга как причины комы большую роль играет исследование давления цереброспинальной жидкости (при спинальной пункции), которое отражает величину внутричерепного давления [Салалыкин В.И., Арутюнов А.И.,. 1978]. В норме давление цереброспинальной жидкости не должно превышать 120—180 мм вод. ст. Повышение его до 400— 500 мм вод. ст. свидетельствует об увеличении внутричерепного давления, которое может быть обусловлено отеком мозга (или каким-либо объемным образованием, например опухолью). В ряде случаев давление цереброспинальной жидкости достигает 800—1000 мм вод. ст.
С клинической точки зрения понятия «отек мозга» и его «набухание» почти равнозначны. Во всяком случае различия между ними определить всегда очень трудно [Двитницкий-Рыжов Ю.Н., 1964; Смирнов А.В., 1969], поэтому клиницисты часто пользуются термином «отек—набухание головного мозга» [Арутюнов А. И., 1960].
Электроэнцефалография. Для оценки функции мозга при коматозных состояниях часто используют электроэнцефалографию (рис. 7.1, 7.2). Поскольку этот метод отражает электрическую активность мозга (и в лучшем случае можно получить представление об ее изменениях в различных зонах мозга), изменения характера ЭЭГ нельзя считать специфичными для комы. Электрокардиографическая картина часто не совпадает с состоянием функциональной активности отдельных структур головного мозга, поэтому нередко можно получить, например, сходные характеристики кривых ЭЭГ при коме наркотического происхождения и при глубокой гипоксической коме. Тем не менее электрокардиография как метод диагностики имеет большое значение в оценке комы, так как облегчает диагностику и способствует оценке динамики комы (см. рис. 7.2). Однако наша практика показывает, что прогностическое значение ЭЭГ при коме невелико.
Это положение может быть иллюстрировано следующим наблюдением.
Больной С., 62 лет, поступил в клинику в связи с желудочным кровотечением. Проведен комплекс мероприятий, направленных на восстановление ОЦК и поддержание кровообращения. После стабилизации состояния выполнена гастроскопия. Установлено наличие на задней стенке желудка гемангиомы, являющейся источником кровотечения. Два года назад больной подвергся оперативному вмешательству в связи с желудочным кровотечением. Операция закончилась гастротомией и прошиванием кровоточащих сосудов. Радикальная операция оказалась невозможной из-за анатомических особенностей и сосудистых аномалий.
При гастроскопии признаков продолжающегося кровотечения не обнаружено. Через 2 сут началось повторное массивное желудочное кровотечение, по поводу которого больной немедленно взят в операционную. В момент транспортировки возникла асистолия и развилась клиническая смерть. Немедленно применен комплекс реанимационных мероприятий, включавший наружный массаж сердца, ИВЛ и последующую массивную гемотрансфузию; кровообращение удалось восстановить. Произведены гастротомия и прошивание конгломерата кровоточащих сосудов на задней стенке желудка.
После операции больной оставлен на продленной ИВЛ, но добиться пробуждения его не удалось. Состояние оценено как коматозное в связи с перенесенной клинической смертью, геморрагическим шоком, гиповолемией, постгеморрагическим ДВС-синдромом. Кома выражалась в полном отсутствии сознания, арефлексии, сопровождалась мышечной гипотонией. Однако наблюдалась нерегулярная спонтанная дыхательная активность, требовавшая медикаментозной синхронизации с использованием мышечных релаксантов. Через 4 сут спонтанное дыхание прекратилось полностью и отпала необходимость в медикаментозной синхронизации. Кома в этот момент оценена как ареактивная (coma cams no Bozza Marrubini). Медленноволновая дельта-активность на ЭЭГ с редкими всплесками ее в виде разрядов, наблюдавшаяся в первые 4 сут, постепенно сменилась полным отсутствием колебаний практически во всех зонах мозга.
В течение 2 нед состояние было крайне тяжелым. На фоне продолжавшейся ИВЛ, интенсивной инфузионной и гемотрансфузионной терапии, противоотечной терапии, а также медикаментозного лечения (применение дофамина, нитролингвала, стероидных гормонов, массивная антибиотикотерапия, иммунокоррекция и др.), СДРВ и тяжелого ДВС-синдрома, почечной недостаточности, метаболического постгипоксического алкалоза многократно возникали эпизоды желудочного кровотечения, всякий раз ухудшавшие общее состояние и усиливавшие проявления полиорганной недостаточности. В результате нескольких попыток удалось эмболизировать чрезаортальным способом конгломерат аномальных артериальных стволов в зоне селезеночной и левой желудочной артерий. Кровотечение прекратилось, общее состояние стабилизировалось, гемодинамика улучшилась. Хотя наркотических анальгетиков и релаксантов в этом периоде больной не получал, он продолжал оставаться в состоянии сверхглубокой комы. Спонтанная дыхательная активность полностью отсутствовала. Зрачковые рефлексы не отмечались, но зрачки были расширены лишь незначительно. При неврологическом исследовании сухожильные и мышечные рефлексы не вызывались. Ежедневно на ЗЭГ фиксировалось практически полное отсутствие электрической активности мозга. Это давало основание, учитывая другие проявления комы, оценивать прогноз как крайне неблагоприятный.
Через несколько дней после полного прекращения кровопотери состояние удалось стабилизировать путем применения сложного комплекса мероприятий, направленных на нормализацию внутренней среды организма, волемии, осмоляльности плазмы, КОС, гемодинамики, выделительных и других функций. Проводилась также борьба с постгипоксическим отеком мозга (диуретические средства, многократные сеансы гемофильтрации). Продолжалась ИВЛ через трахеостому.
Через 1 нед после эмболизации сосудов и полного прекращения кровопотери появились низкие сухожильные рефлексы и слабая двигательная реакция на болевые раздражения. Еще через 2 дня стало проявляться спонтанное дыхание, которое оставалось неадекватным. Этому сопутствовало улучшение электроэнцефалографической картины: появившаяся вначале медленноволновая дельта-активность сменилась а -подобной и вскоре ЭЭГ нормализовалась. Больной начал проявлять элементарные признаки пробуждения — открывать глаза и по инструкции двигать конечностями. Через 42 дня от начала ИВЛ, после недельного периода тренировок спонтанного дыхания удалось полностью отключить респиратор и восстановить адекватную дыхательную функцию. К этому моменту полностью восстановились сознание и «ориентировка в окружающем, но отмечалась выраженная психическая астения.
Через 68 дней больной в удовлетворительном состоянии переведен в санаторное учреждение для окончательной реабилитации.
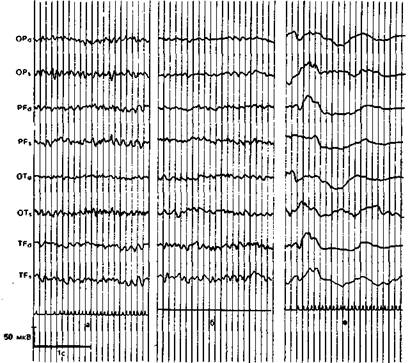
Рис. 7.1. Динамика ЭЭГ (8 отведений) при прогрессирующей коме.
а —исходная ЭЭГ, больной в сознании; б— сознание утрачено, дыхание не нарушено, рефлекторная активность сохранена; в — ареактивная кома, ИВЛ.



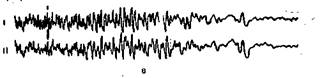


Рис. 7.2. Варианты нарушений ЭЭГ у больных в состоянии прекомы и комы. I — лобно-затылочное; II — теменно-затылочное отведение; а— преобладает бета-активность. Начальные стадии острой печеночной недостаточности. Больной в сознании. беспокоен; б — преобладает а -активность, редкие 8-волны. Острая почечная недостаточность. Больной в сознании заторможен: в — переход бета-активности в преобладающую 6-активность. Развитие токсико-инфекционного шока, кома; г — преобладающая делта-активность с примесью других видов активности; д — непрерывная спайковая активность на фоне 6-активности. Эпизодически появляются зоны сниженной почти до изоэлектрической линии активности длительностью около 1 с и более. Кома, сохранена болевая реакция в виде экстензорных судорог; е — изоэлектрическая линия биоэлектрическая активность практически отсутствует. Ареактивная кома.
Компьютерная томография. С внедрением в клиническую практику аксиальной компьютерной томографии появилась возможность прямой неинвазивной диагностики отека — набухания головного мозга, а также возможность наблюдать его динамику и результаты лечения. Для зон отека мозга характерно снижение плотности мозгового вещества из-за повышения содержания воды в сочетании с расширением желудочков мозга.
7.5. Клинические варианты комы
Постишемическая (постгипоксическая) кома. В настоящее время в зарубежной литературе варианты повреждения мозга и комы, возникающей после прекращения кровообращения (например, в результате остановки сердца), принято называть глобальной церебральной ишемией — аноксией [Safar P. el al., 1978]. Социально-экономическое и клиническое значение этого явления вытекает из того факта, что большая часть больных, реанимированных после остановки сердца, умирают в результате вторично развившейся смерти мозга, а более чем у 20% выживших наблюдаются тяжелые перманентные нарушения мозговой функции [Bell J. A., Hodgson L. Н., 1974]. Пессимистическая концепция относительно возможности восстановления функции мозга и выживания организма после остановки сердца более 4—6 мин в настоящее время считается не оправдавшейся [Гурвич А.М., 1985; Safar P. et al., 1978]. В литературе приводятся свидетельства возможности полного восстановления и выживания даже после более длительной остановки сердца, хотя подобные случаи не часты. По-видимому, это в значительной степени связано с усовершенствованием методов сердечно-легочно-мозговой резусцитации [Safar P. et al., 1978J, применением специальных методов лечения гипоксии мозга, монито-рирования функций и адекватного общего интенсивного лечения.
В результате в клиники и больницы стали периодически поступать больные, которым требуется длительное и сложное лечение в связи с постгипоксической комой и отеком мозга, сопровождающимися недостаточностью других функций.
Изменения в мозге после остановки кровообращения, прекращения дыхания и применения методов реанимации всегда многообразны и комплексны. Они являются результатом сложения последствий первичного «глобального инсульта» и вторичных постишемических изменений, которые включают в себя динамическую многофокусную гипоперфузионную или гипоксическую гипоксию и гиперметаболизм мозга [Fischer E., 1973; Lind В. et al., 1975].
При внезапном тотальном прекращении мозгового кровообращения уже через 10 с истощаются запасы О2 в мозге и наступает потеря сознания. Снижение уровня глюкозы, а также прекращение низкоэнергетического анаэробного метаболизма происходит в течение последующих 4—6 мин. Затем окончательно исчерпываются запасы высокоэнергетических фосфатов (АТФ). В результате в течение 5 мин прекращаются все процессы, требующие энергии, что приводит к прекращению специфической деятельности нейронов. Дефицит внутриклеточной энергии обусловливает прекращение деятельности натриевого насоса, задержку Na+ в клетке и возникновение вследствие этого внутриклеточной гиперосмоляльности и отека (набухания) клетки. Несколько позднее нарушается также функция гематоэнцефалического барьера, что приводит к интерстициальному отеку мозга и множественным микрогеморрагиям [Hossmann К.A., Kleihenes P., 1973].
Важно подчеркнуть, что общего гиперосмоляльного синдрома (по осмоляльности плазмы крови) на первых этапах развития постишемической комы, как правило, не бывает. Он может развиться на более поздних этапах заболевания, когда присоединяется полиорганная недостаточность (например, почечная недостаточность и задержка натрийуреза или азотемия) или возникают ятрогенные изменения содержания Na+ в плазме. Все описанные нарушения водного баланса в мозге, обусловливающие его отек, связаны с клеточной гиперосмоляльностью, которая обычно не отражается на содержании Na+ в плазме.
Полная остановка кровообращения в течение 5—7 мин приводит к многочисленным гистологическим изменениям, включающим рассеянный некроз большого числа нейронов.
Возобновление кровообращения через 5—10 мин после его прекращения приводит к гиперемии мозга и последующему повышению внутричерепного давления. Однако вследствие ДВО синдрома, сладжирования клеток крови и увеличения размеров, нейронов и эндотелиальных клеток общий объем мозгового кровотока бывает низким. Регуляция мозгового кровотока через изменения Рсо2 прекращается. Таким образом, повышение внутричерепного давления, как правило, развивающееся через 30—60 мин после возобновления кровообращения, является: следствием увеличения критической массы мозга и снижения мозгового перфузионного давления.
При недостаточности мозгового кровообращения (среднее артериальное давление около 50 мм рт. ст.), т. е. при неполной временной ишемии мозга, сохраняется ауторегуляция мозгового кровотока, временно поддерживающая мозговое кровообращение на удовлетворительном уровне при сохраненном сознании.
Если среднее артериальное давление продолжительное время: остается на уровне ниже 50 мм рт. ст., то может возникнуть делирий или кома [Safar P., 1983]. При среднем артериальном давлении ниже 30 мм рт. ст. мозговой кровоток составляет не более 20% нормального и выживание нейронов становится сомнительным. Однако депрессия ЦНС (по неврологическим признакам) не коррелирует с ОЦК и СВ. Достаточно хорошая: корреляция прослеживается между депрессией ЦНС и уровнем мозгового перфузионного давления крови, т. е. по разности среднего артериального и внутричерепного давлений.
В случае сопутствующей гипоксемии снижение транспорта О2 может оказаться критическим, даже если гипотония умеренная.
Таким образом, вторичная гипоксемия (или травма) может сделать продолжительный гиповолемический шок необратимым. В то же время поддержание адекватного мозгового кровообращения существенно увеличивает вероятность выживания при шоке [Kovach A., Sandor P., 1976].
Нормальный мозг более устойчив к гипоксемии, чем к ишемии. Если РаО2 снижается до 30 мм рт. ст. или даже ниже в условиях нормального перфузионного давления, то в мозге развивается лактат-ацидоз. Некроза нейронов в этих условиях не происходит, если не развивается дальнейшее снижение РаО2 или если не присоединяется гипотензия [Safar P., 1983].
Гипоксемия практически всегда имеет место у больного в коматозном состоянии. Она может возникать вследствие аспирации (или какой-либо другой причины перенесенной больным асфиксии), шокового легкого или подавления центральной регуляции дыхания из-за отека мозга. У ряда больных возникает нейрогенный отек легких, природа которого мало изучена. Предположительно нейрогенный отек легких может развиться вследствие массивного выброса катехоламинов, приводящего к переполнению легочного сосудистого русла. Этому способствуют внутричерепная гипертензия, гипоксия мозга и нарушение функции гипоталамуса [Thedore J., Robin E., 1975].
Лечение. Этиологическая неспецифичность коматозного состояния, возникающего в большинстве случаев как результат анатомического нарушения клеточных структур мозга или как следствие глубоких расстройств метаболизма клеток, например гипоксии, обусловливает необходимость синдромного принципа ведения коматозного больного. Предусматривается одновременная или поэтапная коррекция основных соматических расстройств. Бесполезно, например, пытаться медикаментозными средствами бороться с комой, если не устранен СДРВ, определяющий гипоксемию, или гипергликемия при диабетической коме.
В связи с этим целесообразно привести примерный перечень лечебных мероприятий общего характера. По-видимому, они должны проводиться в следующей последовательности.
1. Предупреждение обструкции дыхательных путей и обеспечение эффективности дыхания (использование различных положений тела или применение воздуховодов, туалет глотки и трахеи, увлажнение дыхательной смеси и т. д.).
2. Точное определение степени оксигенации крови и оценка эффективности оксигенирующей функции легких. При оценке степени кислородной недостаточности важно установить не столько абсолютный уровень оксигенации артериальной крови, сколько зависимость оксигенации от кислородного режима: если при спонтанном дыхании 100% кислородом Ра02 не превышает 100 мм рт. ст., то больной нуждается в ИВЛ в связи с СДРВ.
Для ИВЛ используют смеси О2 (50—60%) с воздухом.
Если ИВЛ предполагается как кратковременная мера поддержания оксигенации организма, то можно ограничиться назо- или оротрахеальной интубацией, но при длительной ИВЛ целесообразна трахеостомия.
Установление точной причины комы (обследование больного) и последующие лечебные мероприятия, в том числе оперативное вмешательство, целесообразны лишь после того, как обеспечена эффективность дыхания и кровообращения.
3. Мониторирование и поддержание оптимального уровня системного кровообращения как единственная возможность обеспечить адекватный мозговой кровоток. Возможны различные методы мониторирования и ориентировочного обследования: от контроля артериального давления (оптимальное среднее 90— 110 мм рт. ст.) и ЦВД до определения СВ, внутрисердечного давления и ОЦК. Дефицит ОЦК корригируют путем инфузии плазмоэкспандеров, плазмы и гемотрансфузии. Нормоволемические варианты гипотензии требуют лечения дофамином. При гипертоническом кризе показано применение 150—300 мкг клонидина (гемитона).
4. Поддержание нормального водно-электролитного и белкового баланса организма. Важно подчеркнуть, что нарушение содержания электролитов и концентрации протеинов в плазме крови, определяющих осмоляльность внеклеточной среды и КОД плазмы, является предпосылкой отека интерстициального пространства, а затем и набухания клеток мозга. Необходим также контроль уровня шлаков в крови. Накопление мочевины способствует увеличению осмоляльности (но не тоничности) плазмы и интерстициального пространства и усиливает отек мозга. Борьба с расстройствами водно-электролитного баланса может осуществляться с помощью диуретических средств, путем оптимизации водной нагрузки и выбора инфузионных сред, а также применением ультрафильтрации плазмы и гемофильтрации.
В ряде случаев расстройства электролитного баланса у больных в коматозном состоянии развиваются стремительно. Иногда это выражается в существенном повышении уровня Na+ в плазме. Мы наблюдали больных, у которых концентрация Na+ плазмы достигала 180—186 ммоль/л при осмоляльности плазмы выше 400 мосмоль/кг. Прирост осмоляльности внеклеточной среды выше двойной концентрации Na+ был обусловлен, как правило, высоким уровнем мочевины и сахара крови, суммарная концентрация которых в ряде случаев составляла 40— 60 ммоль/л.
Основные патогенетические механизмы повышения концентрации Na+ в плазме связаны, с одной стороны, с задержкой, Na+ на уровне почек у больных в критическом состоянии, с другой — с перегрузкой организма Na+. При условии ограничения или полного исключения введения натриевых растворов (изотонический раствор хлорида натрия, декстраны в сочетании с Na+, растворы аминокислот, содержащие Na+, раствор Рингера—Локка и др.), введение которых в условиях задержки натрия совершенно обоснованно представляется нецелесообразным, главными источниками поступления Na+ в организм (помимо других, менее значительных) являются растворы оксибутирата натрия и натриевых солей антибиотиков. Как правило, эти источники ускользают от внимания врача и повышение концентрации Na+ в крови нередко вызывает у него недоумение. Между тем больной в состоянии комы, находящийся на ИВЛ, может получить в сутки до 20—40 г оксибутирата натрия и несколько граммов антибиотиков. При нормальной функции почек и нормальном натрийурезе этот избыток Na+ быстро элиминируется. При скрытой почечной недостаточности, вторичном постстрессовом альдостеронизме избыток Na+ в организме задерживается.
В последние годы пересматриваются позиции относительна целесообразности традиционного и принятого во всем мире внутривенного введения гидрокарбоната натрия в схеме классической сердечно-легочной реанимации. Н. P. Muster (1987) рекомендует воздерживаться от применения этого препарата, даже если ацидоз документирован. Автор установил, что в тех случаях, когда осмоляльность плазмы выше 350 мосмоль/кг, из числа больных, получивших гидрокарбонат натрия, после пережитой сердечно-легочной реанимации погибает 90%. Вот почему мы настаиваем на применении различных по временным интервалам вариантов мониторирования уровня электролитов плазмы у больных в критическом состоянии, поскольку борьба с отеком мозга и комой невозможна, если развился гиперосмоляльный синдром.
У некоторых больных целесообразно контролировать электролиты плазмы крови дважды в сутки и определять экскрецию их в суточной моче.
Мы надеемся, что после знакомства с элементами этой проблемы читатель понимает также вредоносный эффект внутривенного введения гипертонических растворов для так называемой борьбы с отеком мозга, столь широко применявшийся в недавние годы. Больной в состоянии комы нуждается прежде всего в нормализации водно-электролитного баланса методами, упомянутыми выше. Лишь в отдельных случаях следует вводить белковые препараты (альбумин, плазма) для повышения КОД. Однако если при этом уровень альбумина и белков крови в целом не повышается (по крайней мере на половину введенной дозы белков), то это значит, что поражение сосудистой стенки в ходе развития критического состояния и полиорганной недостаточности уже привело к нарушению функции удержания белка, в результате чего сосуды стали патологически проницаемы для белков. Это соответственно ведет к усилению интерстициального отека мозга, легких и других органов. В подобных случаях лечение с использованием белковых препаратов целесообразно прекратить.
5. Поддержание нормальной температуры тела. Применяют антипиретики (например, ацетилсалициловую кислоту в дозе 10—20 мг/кг), вазодилататоры (аминазин по 0,2—0,5 мг/кг), поверхностное охлаждение.
6. Седативная терапия. При двигательном возбуждении целесообразно вводить внутривенно бензодиазепины (диазепам, седуксен, валиум) в дозе 0,1—1,0 мг/кг. При судорожном припадке вводят барбитураты (5—10 мг/кг), при экстензорных спазмах — бензодиазепины.
Если после введения барбитуратов и бензодиазепинов судороги и спазмы не прекращаются, то применяют миорелаксанты.
7. Синхронизации дыхательной активности больного с аппаратом достигают по обычной методике, предусматривающей гипервентиляцию, последовательное применение бензодиазепинов, оксибутирата натрия, морфина, фентанила, барбитуратов и кураризации.
8. Около 10 лет назад школой P. Safar в Питтсбурге [В1еyaert A., et al., 1978] экспериментально и клинически изучен эффект внутривенного введения барбитуратов при постишемической (постгипоксической) коме. Установлено, что при этих состояниях барбитураты снижают уровень метаболических процессов в мозге и уменьшают выраженность отека мозга, подавляют судорожную активность и тем самым повышают шансы на выживание. Кроме того, благодаря анестезирующему эффекту барбитураты снижают разрушающее влияние ноцицептивной стимуляции. Наблюдаются и другие эффекты, которые, однако, недостаточно документированы [Safar P., 1983]. Среди них называют способность барбитуратов связывать и элиминировать свободные радикалы, образовавшиеся во время ишемии Demopoulos Н. В. et al., 1977], изменять метаболические пути, подавлять катехоламинобусловленный гиперметаболизм [Nemoto E., 1987] и др.
Целесообразно вводить барбитураты (тиопентал-натрий) по 10—15 мг/кг через 10—50 мин по восстановлении кровообращения после асистолии. Затем в течение 1 сут тиопентал-натрий вводят капельно в дозе 20 мг/кг. Однако имеется определенный риск влияния барбитуратов на сердечную деятельность вплоть до повторной асистолии [Breivik H. et al., 1978].
9. Борьба с отеком мозга. Одним из элементов борьбы с отеком и оптимизации метаболизма мозга принято считать стероидную терапию. В настоящее время практически во всех случаях коматозного состояния применяют дексаметазон [Cooper P. et al., 1979] по 0,4 мг/кг в 1-е сутки и 0,2 мг/кг в сутки в последующие 6 дней. Иногда используют дексаметазон в так называемых мегадозах (2,5 мг/кг в сутки первоначально и по 1,5 мг/кг в сутки в последующие 6 дней) [Gudeman S. et al., 1979] или гидрокортизон в больших дозах (до 1 г/сут).
Хотя кортикостероиды почти всегда применяют при травме мозга, инсультах, постгипоксической и других вариантах энцефалопатии, некоторые авторы подвергают сомнению протективный и лечебный эффект этих препаратов при коме [Shapiro H., 1975]. Обзор литературы [Cooper P. et al., 1979] и анализ статистических данных позволили сделать вывод, что применение кортикостероидов не меняет существенно течения ишемического, цитотоксического или вазогенного отека мозга. Однако остается неясным вопрос об эффективности больших доз глюкокортикоидов.
Весьма заманчивой представлялась попытка лечить кому так называемыми пробуждающими средствами — стимуляторами ЦНС, аналептиками и другими активаторами метаболизма мозга, такими как кордиамин, налорфин, меклофеноксат или цитидин дифосфохолин. Однако заметных результатов в этом направлении не получено [Bozza-Marrubini M., 1983]. В клинической практике не оправдало надежду также использование предшественника дофамина L-ДОФА при длительной коме или апаллическом синдроме. Физостигмин как ингибитор холинэстеразы, проникающий через гематоэнцефалический барьер, оказался достаточно эффективным при лечении комы, галюцинаторных состояний и патологической двигательной активности,, вызванных передозировкой атропина (или других препаратов белладонны), фенотиазиновых препаратов, трициклических антидепрессантов, антигистаминных препаратов.
При коме и отеке мозга ИВЛ является одним из главных принципов лечения. По мнению G. Marrubini (1966), применение ИВЛ существенно снизило смертность больных в коматозном состоянии. Считается целесообразным поддерживать РаO2 на уровне выше 100—120 мм рт. ст. с умеренной гипокапнией. (Р a со2 — 25—30 мм рт. ст.), т. е. целесообразна умеренная гипервентиляция.
Одним из важных элементов борьбы с отеком мозга и, следовательно, с комой является диуретическая терапия фуросемидом, который дозируют в зависимости от эффекта, обычно по 40—60 мг, а при поражении почек и задержке мочи до 400—800 мг в сутки. При вторичном гиперальдостеронизме применение салуретиков целесообразно дополнять альдактоном в дозе 400—600 мг в сутки внутривенно или верошпироном внутрь, В отдельных случаях в отсутствие гиперосмоляльного синдрома используют осмодиуретики: маннитол по 1—2 г/кг (20—25% раствор), сорбитол по 2—3 г/кг (20—30% раствор) и глицерин, по 1—2 г/кг.
Маннитол — препарат шестиатомного спирта маннита. Он практически не проникает через гематоэнцефалический барьер и другие клеточные мембраны и в связи с этим повышает осмоляльность крови. Маннит хорошо фильтруется в почках в неизмененном виде и почти не реабсорбируется в канальцах. Именно на этом основан его осмодиуретический эффект. В отличие от мочевины (которая также является мощным осмодиуретиком, но не может быть рекомендована для лечения коматозных состояний и отека мозга) маннитолу почти не свойственны феномен отдачи и вторая волна отека и набухания мозга после окончания диуретической фазы его действия.
Сорбитол — препарат сорбита, применяемый в виде гипертонических растворов (10—30%), вызывает усиление диуреза, хотя и менее выраженное, чем маннитол. В отличие от маннитола сорбитол метаболизируется в организме как сахар с продукцией энергии эквивалентно глюкозе.
Глицерин — трехатомный спирт — повышает осмоляльность плазмы и благодаря этому обеспечивает дегидратирующий эффект. Глицерин нетоксичен, не проникает через ГЭБ и в связи с этим не вызывает феномена отдачи.
Фуросемид (лазикс) и этакриновая кислота являются достаточно мощными салуретическими препаратами, оказывают диуретическое и, следовательно, дегидратирующее действие путем торможения реабсорбции Na+ и Cl— в канальцах почек. Важным качеством салуретиков является их способность снижать продукцию цереброспинальной жидкости [Bozza-Marrubini M., 1983].
Следует подчеркнуть, что эффективное лечение комы, связанной с отеком мозга, достигается скорее путем устранения метаболических расстройств, вызвавших отек мозга, чем медикаментозной ликвидацией самого отека мозга. Эта задача не может быть успешно решена, если не нормализованы кровообращение, окислительные процессы, водно-электролитное и кислотно-основное состояние, а также выделительная и другие функции организма.
Кома при черепно-мозговой травме. Повреждения черепа и мозга являются одной из основных причин коматозных состояний. В 1975 г. в США зарегистрировано 633000 случаев тяжелых травм черепа, в результате которых 60% пациентов умерли или остались живыми с серьезными необратимыми неврологическими функциональными расстройствами. Подавляющее большинство из них пережили кому различной выраженности. Основными видами повреждений явились частичное или распространенное разрушение вещества мозга, ушиб его, субдуральные и эпидуральные гематомы [Bruce D. et al., 1978].
Клиническая картина коматозного повреждения при травме мозга весьма многообразна и характеризуется сочетанием локальных и общих неврологических симптомов.
Анамнез, не только объясняющий механизм травмы и ее обстоятельства, но и позволяющий оценить развитие постравматического состояния, имеет большое значение. Если, например, больной к моменту прибытия бригады скорой помощи на место происшествия был в сознании, а во время транспортировки в клинику впал в коматозное состояние с потерей сознания, то есть основания подозревать внутричерепное кровоизлияние или прогрессирующий отек мозга. Провести дифференциальную диагностику помогает неврологическое обследование. При выявлении неврологической асимметрии следует предполагать гематому.
В клинических условиях прежде всего целесообразно оценить состояние больного с использованием шкалы комы Глазго. Оценка меньше 8—7 баллов свидетельствует о тяжелом повреждении мозга. Неврологическое обследование должно быть обстоятельным, точным, быстрым и не должно мешать проведению интенсивной терапии. Нельзя, например, задерживать введение мышечных релаксантов для последующего подключения ИВЛ у больного с расстройствами дыхания только потому, что предстоит исследование сухожильных и мышечных рефлексов.
Если больной в коме не открывает глаз по команде и не проявляет реакций на речевое обращение, то целью обследования должно быть выяснение степени дисфункции коры мозга, диэнцефальных и мезэнцефальных отделов мозга, моста мозга и, определение односторонних неврологических дефицитов, указывающих на наличие гематомы. В рамках первого клинического-обследования обязательна поясничная пункция. Кровь в цереброспинальной жидкости указывает на внутричерепное кровоизлияние, высокое давление, отсутствие крови — на уже развившийся отек мозга. При установлении диагноза внутричерепной гематомы вопрос о показаниях к оперативному ее удалению-решается в зависимости от степени нарушения функций мозга и угрозы ущемления мозга в тенториальном отверстии.
Повторное неврологическое обследование осуществляют через 2 ч. Иногда оно имеет большее значение, чем первоначальное обследование, так как позволяет выявить динамику состояния и окончательно определить дальнейшую тактику ведения — консервативную или оперативную.
Важной частью обследования является оценка уровня сознания, определяемая по двигательным реакциям, диаметру зрачков, реакции их на свет, корнеальным рефлексам, глазодвигательным функциям.
Функциональное состояние ствола мозга и нижних отделов; моста мозга можно оценить по состоянию спонтанного дыхания (см. выше) и наличию или отсутствию «рефлекса кляпа».
Наличие мышечного гипертонуса по типу декортикационной или децеребрационной ригидности или, наоборот, мышечной атонии в сочетании с равномерным максимальным расширением зрачков — неблагоприятные прогностические признаки.
Как и при постишемической коме, на первых этапах развития посттравматической комы общего гиперосмоляльного синдрома не бывает.
Элементы лечения. В дополнение к общей программе лечения больного в коматозном состоянии, изложенной в разделе «Постишемическая кома», необходимо подчеркнуть следующее.
1. Всем больным, которые не реагируют на обращение и болевые раздражения, должна быть проведена интубация трахеи. Рекомендации ранней интубации основаны на высокой смертности больных с повреждением мозга и в состоянии комы, которая бывает обусловлена главным образом гипоксемией [Becker D., Gudeman S., 1980]. Недостаточность дыхания при коме может развиться внезапно, чаще всего в связи с обструкцией дыхательных путей или с ущемлением мозга. Если необходима компьютерная томография мозга, то обязательна иммобилизация тела, которой можно достичь лишь с помощью миорелаксантов и, конечно, интубации трахеи. С этой целью предпочтительно применять панкурониум бромид (павулон).
2. Необходима по возможности ранняя коррекция артериального давления. В отдельных случаях при коме может восстановиться сознание, если артериальная гипотензия корригирована. Результаты неврологического обследования могут быть ошибочными, если артериальное давление на уровне 40—50 мм рт. ст. Однако надо помнить; что коррекция гипотензии в подобных случаях — процедура чрезвычайно тонкая и нередко опасная, ибо у ряда больных поспешная и неосторожная нормализация артериального давления, особенно если неправильно выбран метод (например, с использованием катехоламинов), может привести к катастрофическому повышению внутричерепного давления с последующим резким ухудшением состояния. Коррекцию гипотензии нельзя осуществлять до интубации трахеи и перевода больного в коме на ИВЛ. Вместе с тем продолжительная гипотензия сама по себе опасна, так как ухудшает кровообращение в мозге и снижает шансы на выживание. Методы коррекции гипотензии зависят от ее этиологии. При гиповолемическом (постгеморрагическом) ее варианте применяют гемотрансфузию, переливание плазмы, белковых растворов, плазмоэкспандеров. При сосудистом и кардиальном варианте вводят дофамин.
3. Если отмечается ацидоз, то чаще всего он лактатный и, следовательно, требует коррекции гидрокарбонатом натрия. Однако доза этого препарата должна быть ограничена (см. выше) 1 ммоль/кг (из-за опасности перехода в алкалоз и развития гиперосмоляльного синдрома) и может вводиться лишь при удовлетворительной функции почек. Предварительно следует убедиться в том, что ацидоз не связан с гипоксией и гиперкапнией.
4. Все больные в коме, обусловленной травмой черепа и повреждением мозга, должны быть консультированы нейрохирургом, который прежде всего должен определить показания к оперативному вмешательству (например, в связи с внутричерепной гематомой) или подтвердить отсутствие таковых.
5. Лечение отека мозга можно проводить только после установления факта отсутствия внутричерепной гематомы, ибо при наличии ее подобное лечение будет безрезультатным и даже вредным.
В остальном лечение осуществляют по принципам, описанным в разделе «Постишемическая кома».
Кетоацидотическая (диабетическая) кома. Представляет собой классический вариант комы при сахарном диабете и проявляется потерей сознания в результате полного исчерпания запасов эндогенного или экзогенного инсулина и перехода метаболизма на липидный путь с образованием большого количества бета-гидроксибутирата, ацетоацетата и ацетона (так называемых кетоновых тел), уровень которых повышается (до 3— 5 ммоль/л) в крови, а затем и в моче.
Плазменная гипергликемия, обусловливающая гиперосмоляльный синдром, вызывает осмодиурез, дегидратацию, вторичную гипонатриемию и гипокалиемию. У больных развиваются типичная одышка (дыхание Куссмауля), гипотензия и ацидоз. В моче определяются кетоновые тела, от больного пахнет ацетоном. Потери воды организмом иногда составляют 5—8 л, Na+ и К+ — по 300—500 ммоль. При кетоацидотической коме водные и электролитные потери почти всегда выше, чем при некетотических формах. Гиперосмоляльный синдром способствует возникновению отека мозга. Концентрация сахара в крови повышается до 22—55 ммоль/л (4—10 г/л), в моче — до 220— 250 ммоль/л.
Довольно быстро развивается депрессия ЦНС, переходящая в коматозное состояние с многообразной неврологической картиной. Возникают делирий, общее возбуждение, судорожный синдром, повышается рефлекторная активность. Выраженная одышка иногда сопровождается расстройствами ритмики дыхания.
Лечение гипергликемической диабетической комы представляет собой весьма сложную задачу прежде всего из-за неблагоприятной комбинации гипергликемии с дегидратацией. Больному немедленно вводят внутривенно 50 ЕД и подкожно 100 ЕД кристаллического инсулина, а затем в течение 1-х суток каждые 2 ч по 25—40 ЕД. В отсутствие быстрого эффекта инсулин должен быть введен внутривенно повторно. Каждые 3 ч контролируют содержание сахара в крови и моче. Каждые 4 ч определяют кетоновые тела в моче. При резком снижении глюкозурии и ацетонурии дозу инсулина снижают до поддерживающей (10—12 ЕД). Одновременно с лечением инсулином начинают внутривенное капельное введение изотонического раствора хлорида натрия вместе с 5% раствором глюкозы и раствором хлорида калия. Общая доза жидкостей должна составлять 3,5—6 л/сут. При развитии сопутствующего лактат-ацидоза [Toker P., 1974] целесообразно ввести внутривенно 250—300 мл 4,5% раствора гидрокарбоната натрия.
Гиперосмоляльная кома может развиться при осмоляльности плазмы выше 325—350 мосмоль/кг [Jimmis H., 1983] и наиболее часто бывает обусловлена гипернатриемией или сочетанием гипернатриемии с гипергликемией.
Происхождение гипернатриемии обусловлено в этих случаях неравномерной потерей Na+ и воды, возникающей в результате глюкозурии при гипергликемическом синдроме. Сверхпотери воды при этом частично возмещаются перемещением воды интерстициального пространства в плазму.
Поскольку гиперосмоляльный синдром часто является результатом сочетания высокой концентрации Na+ и глюкозы, в случае отсутствия осмометра целесообразно оценивать этот суммарный эффект по относительному показателю — фактору осмоляльности (F). Последний может быть вычислен по формуле:
Р = Истинный Na+- [140- (Истинная глюкоза-5)/2 ] ммоль/л
Нормальная величина фактора осмоляльности близка к 5, однако в крайне тяжелых ситуациях она может достигать 40—50 ммоль/л. Еще проще гиперосмоляльность может быть выявлена с учетом удвоенной концентрации Na+ в плазме [Hockaday Т., 1983]. В норме она не должна превышать 270— 280 ммоль/л. При повышении этого показателя до 310— 320 ммоль/л прогноз комы, как правило, плохой. Если имеются данные о концентрации в крови глюкозы, мочевины и белков плазмы, то можно рассчитать осмоляльность плазмы по формуле, приведенной в главе 1.
Гиперосмоляльная кома проявляется беспокойством больного, делирием, мышечной гипертонией, активацией рефлексов, миоклонией, судорогами и нарушением или потерей сознания. Гипергликемическая некетотическая кома (и связанная с нею депрессия ЦНС) возникает наиболее часто у больных сахарным диабетом и имеет множество причин, в основе которых лежит недостаточная коррекция диабета.
Гипергликемическая некетотическая кома в целом выражается клинически как гиперосмоляльный синдром и характеризуется смертностью более 40% [Timmis H., 1983]. При этом наблюдаются выраженный осмодиурез и глюкозурия, резко повышается концентрация сахара в крови (до 30—100 ммоль/л). Гиперосмоляльная кома не сопровождается кетоацидозом, поскольку развивается лишь у больных диабетом с частично сохранившимся островковым аппаратом. При этом сохранившееся небольшое число бета-клеток обеспечивает выделение некоторого количества инсулина, достаточного для торможения липолиза и предупреждения кетоза, но недостаточного для устранения высокой гипергликемии.
Одной из частых причин гиперосмоляльного состояния (в том числе некетотической гиперосмоляльной комы) может быть нарушение секреции альдостерона, приводящее к задержке Na+. Это нередко наблюдается у больных бронхогенным раком. В послеоперационном периоде гиперосмоляльность возникает в результате неправильно сбалансированного зондового питания при недостатке свободной воды в питательной смеси или при гипералиментационном варианте парентерального питания.
Лечение гиперосмоляльной комы заключается в устранении дегидратации и гиповолемии и восстановлении нормальной осмоляльности плазмы. Для этого внутривенно вводят главным образом гипотонические растворы, например 0,45% раствор хлорида натрия, а также 2,5% раствор фруктозы или (в крайнем случае) глюкозы. Иногда для этого необходимо вводить внутривенно гипотонические растворы в дозе до 10—15 л/сут. Важно контролировать уровень К+ в плазме и своевременно корригировать его. Дозу инсулина подбирают индивидуально; в общем она должна быть меньше, чем при лечении диабетической комы.
Гипогликемическое состояние как причина тяжелых функциональных расстройств ЦНС и, следовательно, комы встречается нечасто. Обычно в результате гипогликемии возникает легкое нарушение сознания, которое быстро купируется с началом терапии. Наиболее частая причина гипогликемии (как в клинических, так и в домашних условиях) — передозировка инсулина. Однако гипогликемия может развиваться как вторичное состояние при печеночной недостаточности, когда в крови циркулирует повышенное количество эндогенного инсулина или инсулиноподобного фактора. Уровень сахара при гипогликемии может снижаться до 2,2—1 ммоль/л. Глюкозурии и ацетонурии при этом состоянии не наблюдается. Отсутствуют также признаки дегидратации. Лечение гипогликемического состояния заключается в немедленном внутривенном введении 50—70 мл 40% раствора глюкозы. В отсутствии быстрого эффекта такую же дозу вводят повторно. Через 20—30 мин после введения концентрированного раствора глюкозы начинают капельное внутривенное введение 5% раствора глюкозы (до 1 л).
Печеночная кома. Может возникнуть в терминальной фазе острой или хронической печеночной недостаточности любого происхождения (токсического, инфекционного, алкогольного, лекарственного, вследствие механической желтухи, трансфузионного гепатита, цирроза печени и др.). Печеночная кома нередко сочетается с полифункциональной недостаточностью, обусловливающей критическое состояние. Точно причины печеночной комы обычно трудно выяснить, однако известно, что в критических состояниях ее возникновение может быть активировано высоким содержанием аммиака в желудочно-кишечном тракте, обусловленным либо алиментарной нагрузкой, либо кишечным (или желудочным) кровотечением. Однако этот механизм реализуется лишь в условиях предшествующего поражения печени или портокавального анастомоза [McDermot W., 1968]. В настоящее время имеется множество экспериментальных и клинических доказательств того, что аэробный гликолиз повреждается при высоком содержании в крови аммиака и других азотистых субстанций, которые легко проникают через мембраны нейронов. Прямой связи между выраженностью неврологических симптомов и уровнем аммония в крови нет, однако известно, что развитие комы может быть ускорено лекарственными препаратами, высвобождающими аммоний, такими как дериваты тиазидов, а также экспериментальным введением глицина, который распадается на аммиак и мочевину. Седативные препараты, анальгетики и некоторые другие препараты, которые расщепляются в печени, могут провоцировать печеночную кому, если имеется исходное нарушение функции и структуры печени.
Важную роль в происхождении печеночной комы играет нарушение метаболизма нейромедиаторов в связи с расстройством метаболизма их предшественников — аминокислот.
Печеночная энцефалопатия характеризуется спектром неврологических симптомов — от легких проявлений до выраженного делирия и комы. Печеночный флап (медленный тремор) характерен для прекоматозного периода. Эти симптомы можно наблюдать также при энцефалопатии у больных с легочным сердцем, застойными пороками сердца, полицитемией, уремией и др. При более глубоких степенях энцефалопатии могут отмечаться мышечные спазмы, экстензорные рефлексы и децеребрационная ригидность. Иногда наблюдаются локальные симптомы нарушения мозговой функции, преимущественно в виде парезов. У большинства больных с печеночной комой развивается гипервентиляция и дыхательный алкалоз.
Таким образом, основной механизм формирования печеночной комы связан с повышением уровня аммиака в крови и расстройством нейромедиаторной функции, которые, помимо алиментарных и геморрагических причин, могут быть вызваны извращенным метаболизмом таких аминокислот, как триптофан, тирозин, фенилаланин. В значительной степени этот механизм связан с деградацией бактериальных белков в кишечнике. Признаки энцефалопатии появляются тогда, когда содержание аммиака крови повышается до 4 мкг/мл (нормальный уровень 1 мкг/мл).
Лечение печеночной комы должно предусматривать уменьшение поступления белков и аминокислот в организм, механическое очищение кишечника и снижение активности кишечной флоры путем применения бактерицидных антибиотиков, например неомицина. Применяют большие дозы стероидных гормонов. В ряде случаев такое лечение способствует восстановлению сознания. Важным терапевтическим элементом является использование различных методов детоксикации, направленных на удаление токсических метаболитов, — гемосорбции, плазмафереза, в ряде случаев обменной гемотрансфузии, гемодиализа [Merino G. et al., 1977; Denis L. et al., 1978]. Однако применять эти методы целесообразно лишь тогда, когда имеется обоснованная надежда на восстановление печеночной функции и регенерации печени при обязательном условии ликвидации первоначальной причины ее поражения.
Уремическая кома. Представляет собой одну из последних фаз развития или исход нелеченой или неадекватно леченной острой или хронической почечной недостаточности. В большинстве случаев уремическая кома развивается как результат накопления в организме токсических метаболитов, в частности мочевины, креатинина и других азотистых шлаков, задержки в организме Na+, K+, воды и развития гиперосмоляльного синдрома. Это происходит, если не принимают никаких радикальных мер по выведению шлаков, электролитов и воды, т. е. не проводят гемодиализ. На фоне нарушения жидкостного и электролитного гомеостаза, метаболического ацидоза и артериальной гипертензии развиваются нарушение сознания и неврологические дисфункции, переходящие позже в коматозное состояние. Однако, так же как при печеночной коме, нет четких корреляций между выраженностью коматозного состояния и уровнем азотистых шлаков в крови. Клиническая картина характеризу ется делирием, неадекватным поведением, переходящим затем в ступор и кому, развивающиеся на фоне судорог и локальных неврологических симптомов, таких как асимметрия сухожильных и мышечных рефлексов, иногда гемипареза.
У больных в критических состояниях острая почечная недостаточность, приводящая к уремической коме, в ряде случаев может иметь ятрогенную основу. «Виновниками» ее возникновения в отдельных случаях могут быть гентамицин и другие ами-ногликозиды, колимицин, полимиксин В.
Единственным эффективным методом лечения уремической комы является гемодиализ. Перитонеальный диализ, как правило, малоэффективен. Абсолютным показанием к гемодиализу является уровень азота мочевины выше 30—43 ммоль/л. При возникновении тетанических судорог вводят 5—15 мг диазепама внутривенно. Остальное лечение симптоматическое и должно быть направлено на коррекцию системных дисфункций.
Таким образом, основные варианты коматозных состояний, наиболее часто встречающиеся в клинической практике, имеют существенные патогенетические и клинические различия и в связи с этим требуют различных терапевтических подходов. В табл. 7.4 отражены эти различия по минимальному числу основных патогенетических и диагностических признаков. Мы рассчитываем, что таблица облегчит дифференциальную диагностику основных вариантов коматозных состояний.
Таблица 7.4. Дифференциально-диагностические признаки некоторых коматозных состояний
| Вариант комы | Гиперосмоляль-ный синдром | Гипервентиляция | Дегидра-тация | Кетоз | Концент-рация Na+ плазмы | рН |
| Постишемическая с лактат ацидозом | — | + + + | + | + | N | ↓ |
| Посттравматичеокая | — | + + | + | — | N | ↓ |
| Кетоацидотическая (диабетическая) | + + + + | + + + | + + + + | + + + + | ↓ | ↓ |
| Некетотическая обычная | + + + + | — | + + + + | — | ↓ | N ↓ |
| Гиперосмоляльная | + + + + | — | + + + + | — | ↑ | N |
| Уремическая | + + + | + + | — | — | N или ↑ | ↓ |
| Печеночная | — | — | — | — | К | ↓ |
| Поражение ствола мозга | — | + + + | + | — | N | ↑ |
| Гипогликемическая | — | — | — | — | N | N |
Примечание. Знак «+> — степень выраженности признака, «—«— отсутствие признака, N — норма.