2015-06-10
2015-06-10 1829
1829Несмотря на многочисленные экспериментальные и клинические исследования энцефалопатии, полная картина остаётся во многом противоречивой и спорной. Из имеющихся данных трудно сделать однозначные выводы (табл. 7-4). Важную роль в патогенезе печёночной энцефалопатии играет аммиак, однако другие нейротрансмиттерные системы также вовлечены в патологический процесс.
Таблица 7-4. Трудности при исследовании нейромедиаторов у больных с печёночной энцефалопатией
Доступ к тканям мозга Лабильность факторов, например NH3 Сложность нейромедиаторных систем Проблематичность применения моделей, полученных на животных Значительный спектр заболеваний, характерных для человека Трудности интерпретации сведений, полученных о лигандах, которые зависят от: высвобождения метаболизма (ферментов) выведения/обратного захвата связывания с рецепторами
АММИАК И ГЛУТАМИН
В патогенезе печёночной энцефалопатии аммиак представляет собой наиболее хорошо изученный фактор. Существует множество данных, свидетельствующих о его связи с наблюдающимися нарушениями функции нейронов (рис. 7-6) [48].
Аммиак выделяется при расщеплении белков, аминокислот, пуринов и пиримидинов. Около половины аммиака, поступающего из кишечника, синтезируется бактериями, оставшаяся часть образуется из белков пищи и глутамина. В норме в печени аммиак превращается в мочевину и глутамин. Нарушения цикла мочевины (врождённые дефекты, синдром Рейе) ведут к развитию энцефалопатии.
Уровень аммиака в крови повышен у 90% больных с печёночной энцефалопатией. Содержание его в головном мозге также увеличено. У некоторых больных при пероральном приёме солей аммония может вновь развиться энцефалопатия. Исследования предполагают, что у больных циррозом печени повышается проницаемость гематоэнцефалического барьера для аммиака [36].
Сама по себе гипераммониемия связана со снижением проведения возбуждения в ЦНС. Интоксикация аммиаком ведёт к развитию гиперкинетического предсудорожного состояния, которое невозможно приравнять к печёночной коме.
Предполагают, что при печёночной энцефалопатии основные механизмы действия аммиака заключаются в прямом воздействии на мембраны нейронов или на постсинаптическое торможение [69] и в опосредованном нарушении функций нейронов в результате влияния на глутаматергическую систему.
В головном мозге цикл мочевины не функционирует, поэтому удаление из него аммиака происходит различными путями. В астроцитах под действием глутаминсинтетазы из глутамата и аммиака синтезируется глутамин (рис. 7-7). В условиях избытка аммиака запасы глутамата (важного возбуждающего медиатора) истощаются и происходит накопление глутамина. Содержание глутамина и a-кетоглутарата в спинномозговой жидкости коррелирует со степенью печёночной энцефалопатии. Это лишь упрощенное описание сложного комплекса изменений соотношения глутамин/глутамат, выявляемого при печёночной энцефалопатии [48, 50]. Исследования подтверждают, что при этом происходят редукция мест связывания и уменьшение обратного захвата глутамата астроцитами.
Трудно оценить общий вклад аммиака в развитие печёночной энцефалопатии, в особенности потому, что при этом состоянии наблюдаются изменения и в других нейромедиаторных системах. Участие других механизмов в патогенезе энцефалопатии подчёркивается тем, что у 10 % больных
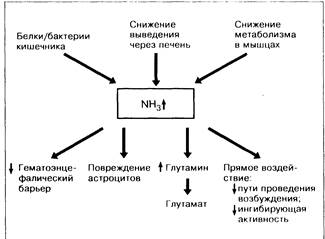
Рис. 7-6. Аммиак: источники образования и возможная роль в развитии печеночной энцефалопатии.
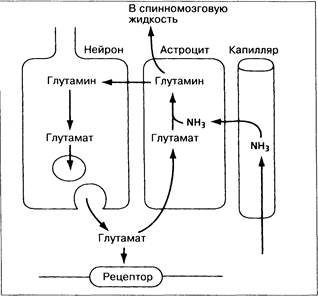
Рис. 7-7. Ключевые этапы глутаматергической синаптической регуляции и выведения аммиака в головном мозге. Глутамат синтезируется в нейронах из своего предшественника глутамина, накапливается в синаптических везикулах и в итоге высвобождается при помощи кальцийзависимого механизма. Высвободившийся глутамат может взаимодействовать с глутаматными рецепторами любого типа, находящимися в синаптической щели. В астроцитах глутамат захватывается и превращается в глутамин при помощи глутаминсинтетазы. При этом используется NH3. Развивающиеся при печёночной энцефалопатии нарушения включают: увеличение содержания NH3 в головном мозге, повреждение астроцитов, уменьшение числа глутаматных рецепторов. (Взято из [48] с разрешения авторов.)
вне зависимости от глубины комы в крови сохраняется нормальный уровень аммиака.
Производные метионина, особенно меркаптаны, вызывают печёночную энцефалопатию. Такие данные привели к предположению, что при печёночной энцефалопатии некоторые токсины, в особенности аммиак, меркаптаны, жирные кислоты и фенолы, действуют как синергисты [79]. Эти наблюдения требуют дальнейшего изучения с использованием более совершенной техники, доступной в настоящее время. По данным последних исследований, при экспериментальной энцефалопатии метанефиол — крайне токсичный меркаптан — не участвует в патогенезе печёночной энцефалопатии [11].
ЛОЖНЫЕ НЕЙРОТРАНСМИТТЕРЫ
Предполагают, что при печёночной энцефалопатии передача импульсов в катехоламиновых и допаминовых синапсах головного мозга подавляется аминами, образующимися под действием бактерий в кишечнике при нарушении метаболизма
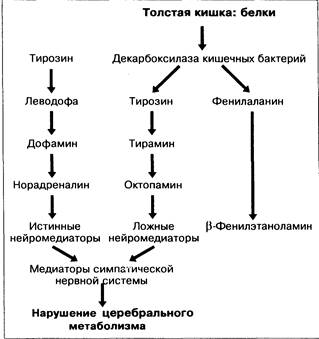
Рис. 7-8. Предполагаемая роль ложных медиаторов симпатической нервной системы в нарушениях церебрального метаболизма у больных с заболеваниями печени.
предшественников нейромедиаторов в головном мозге. В оригинальной гипотезе [22] утверждается, что декарбоксилирование в кишечнике некоторых аминокислот ведёт к образованию b-фенилэтиламина, тирамина и октопамина — так называемых ложных нейротрансмиттеров. Они могут замещать истинные нейромедиаторы (рис. 7-8).
Другое предположение основывается на том, что изменение доступности предшественников медиаторов препятствует нормальной нейропередаче. У больных с заболеваниями печени возрастает содержание в плазме ароматических аминокислот — тирозина, фенилаланина и триптофана, что, вероятно, обусловлено нарушением их дезаминирования в печени. Одновременно понижается содержание аминокислот с разветвленной цепью — валина, лейцина и изолейцина, связанное, вероятно, с увеличением их метаболизма в скелетных мышцах и почках в результате гиперинсулинемии, характерной для больных с хроническими заболеваниями печени. Эти две группы аминокислот конкурируют за прохождение в головной мозг. Нарушение их соотношения в плазме позволяет большему количеству ароматических аминокислот преодолевать нарушенный гематоэнцефалический барьер. При этом состоянии может быть также понижено выведение ароматических аминокислот из головного мозга [29]. Повышение уровня фенилаланина в головном мозге ведет к подавлению синтеза допамина и образованию ложных нейротрансмиттеров: фенилэтаноламина и октопамина.
Улучшение состояния больных при лечении леводофой и бромокриптином подтверждает точку зрения, что при печёночной энцефалопатии наблюдаются изменения в системе нейротрансмиссии, однако число таких больных невелико и результаты неоднозначны. При печёночной энцефалопатии уровень октопамина в сыворотке и моче повышен [38], однако в экспериментах на здоровых крысах внутрижелудочковое введение большого количества октопамина, подавляющее образование в головном мозге допамина и адреналина, не приводило к развитию комы [79]. При посмертном определении содержания катехоламинов в головном мозге у больных циррозом печени с печёночной энцефалопатией их уровень был не ниже, чем у больных циррозом без энцефалопатии на момент смерти [18].
СЕРОТОНИН
Нейромедиатор серотонин (5-гидрокситриптамин) участвует в регуляции уровня возбуждения коры головного мозга и, таким образом, состояния сознания и цикла сон—бодрствование. Предшественник серотонина — триптофан —одна из ароматических аминокислот, содержание которых в плазме повышается при заболеваниях печени. У больных, находящихся в печёночной коме, его уровень в спинномозговой жидкости и головном мозге также повышен; более того, триптофан может стимулировать синтез серотонина в головном мозге. При печёночной энцефалопатии наблюдаются также и другие нарушения метаболизма серотонина, включающие изменения связанных с ним ферментов (моноаминоксидазы), рецепторов и метаболитов (5-гидроксииндолуксусная кислота). Эти нарушения, а также возникновение энцефалопатии у больных с хроническими заболеваниями печени, получавших в связи с портальной гипертензией кетансерин (блокатор 5-НТ-рецепторов) [75], свидетельствуют об участии серотониновой системы в патогенезе печёночной энцефалопатии. Вопрос о том, является ли нарушение в этой системе первичным дефектом, нуждается в дальнейшем изучении.
-АМИНОМАСЛЯНАЯ КИСЛОТА И ЭНДОГЕННЫЕ БЕНЗОДИАЗЕПИНЫ
g-Аминомасляная кислота (ГАМК) представляет собой основной тормозной нейромедиатор в головном мозге. Она синтезируется в пресинаптических нервных окончаниях из глутамата при помощи глутаматдегидрогеназы и накапливается в везикулах. Медиатор связывается со специфическим ГАМК-рецептором на постсинаптической мембране. Рецептор представляет собой часть большого молекулярного комплекса (рис. 7-9), в котором имеются также места связывания с бензодиазепинами и барбитуратами. Связывание любого из этих лигандов ведет к открытию хлорных каналов, после поступления в клетку ионов хлора развиваются гиперполяризация постсинаптической мембраны и торможение нервных импульсов.
ГАМК синтезируется кишечными бактериями, поступает в портальный кровоток и метаболизируется в печени. При печёночной недостаточности или портосистемном шунтировании она попадает в системный кровоток. У больных с заболеваниями печени и печёночной энцефалопатией уровень ГАМК в плазме повышен [34]. Предположение, что ГАМК может участвовать в патогенезе печёночной энцефалопатии, основывается главным
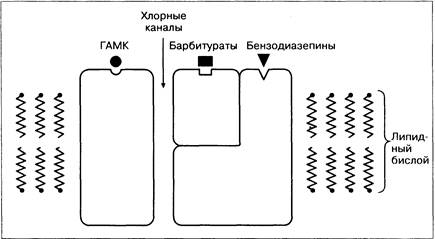
Рис. 7-9. Упрощенная модель комплекса ГАМК-рецептор/ионофор, встроенного в постсинаптическую мембрану нейрона. Связывание любого из изображённых лигандов — ГАМК, барбитуратов или бензодиазепинов — с их специфическими местами связывания ведёт к увеличению прохождения ионов хлора через мембрану. В результате развиваются гиперполяризация мембраны и торможение нервных импульсов [63].
образом на данных, полученных при экспериментальном моделировании острой печёночной недостаточности. Однако результаты исследования головного мозга при циррозе печени с печёночной энцефалопатией на аутопсии не показали роль ГАМК per se в патогенезе энцефалопатии.
Особое внимание к ГАМК-бензодиазепиновому рецепторному комплексу привело к формированию предположения о том, что в организме больных с печёночной энцефалопатией имеются эндогенные бензодиазепины, которые могут взаимодействовать с этим рецепторным комплексом и вызывать торможение. Несмотря на то что при экспериментальной и клинической печёночной энцефалопатии бензодиазепиновые рецепторы не были изменены, у больных с печёночной энцефалопатией, обусловленной циррозом печени, в плазме и спинномозговой жидкости были обнаружены бензодиазепиноподобные соединения [49]; они же были найдены в плазме больных с ОПН [5]. С помощью радиорецепторного анализа показано, что у больных циррозом печени с энцефалопатией, не получавших синтетические бензодиазепины по крайней мере 3 мес, уровень активности бензодиазепинов был значительно выше, чем в контрольной группе обследованных, не имевших заболеваний печени [49|. Тяжесть энцефалопатии коррелирует с бензодиазепиновой активностью плазмы крови и мочи. В кале больных циррозом печени активность бензодиазепиноподобных соединений была в 5 раз выше, чем у обследованных контрольной группы [3].
Остаётся неясным, являются ли изменения в бензодиазепиновых рецепторах или эндогенных лигандах патогенетически значимыми или они представляют собой независимые феномены. Природа лигандов эндогенных бензодиазепиновых рецепторов требует уточнения. Их количество в ЦНС должно быть достаточным для развития энцефалопатии [13]. Тем не менее повышенная чувствительность больных циррозом печени к бензодиазепинам подтверждает участие этой нейромедиаторной системы в патогенезе энцефалопатии [6]. У некоторых больных применение антагониста бензодиазепинов флумаземила приводит к временному (у этого препарата короткий период полувыведения) рефессу печёночной энцефалопатии.