2015-09-06
2015-09-06 7429
74296.1. Электронные состояния и химические связи
в многоатомных молекулах
6.1.1 Характеристика электронных состояний
в многоатомных молекулах
В многоатомных молекулах заметно возрастает число валентных электронов. И задача описания электронных состояний становится более сложной по сравнению с двухатомной молекулой. Если в двухатомной молекуле существует ось симметрии, проходящая через ядра атомов, относительно которой можно рассматривать проекцию орбитального момента электронов и классифицировать этим самым электронные состояния (по проекции орбитального момента на ось молекулы). Для многоатомной молекулы не существует выделенной оси симметрии, и поэтому классификация электронных состояний по типу двухатомной молекулы не применима. Исключения составляют линейные многоатомные молекулы, для которых сохраняется та же классификация электронных состояний, как и для двухатомных (см. гл.5).
Строение электронной оболочки молекулы и в первую очередь ее внешние (валентные) электроны определяют электронный спектр молекулы. Устойчивость электронных состояний определяется свойствами этих состояний по отношению к взаимному расположению ядер в молекуле. В отличие от двухатомной молекулы, в которой осуществляется только одна связь, в многоатомной возникает несколько связей, причем различной силы. Расположение атомов в молекуле и направление химических связей определяет точечную группу симметрии молекулы (равновесную ядерную конфигурацию молекулы). Поэтому электронные состояния многоатомной молекулы могут быть классифицированы по свойствам симметрии. Предполагается, что электронная конфигурация повторяет конфигурацию расположения ядер в молекуле. Если равновесная конфигурация молекулы относится к определенной точечной группе (например, молекула воды относится к точечной группе С 2u), то электронные состояния будем относить к определенным типам симметрии, возможным для данной точечной группы.
Состояния молекул будем классифицировать по свойствам симметрии соответствующих волновых функций. Необходимо выяснить, как ведут себя волновые функции при различных операциях симметрии. Это позволяет их отнести к определенному типу симметрии, а равным образом и те состояния, которые описываются этими функциями. Поэтому все выражения «тип состояния», «симметрия» состояния», «симметрия уровня» энергии должны подразумевать симметрию соответствующих волновых функций, описывающих эти состояния.
Поскольку волновая функция описывает состояние электронов в поле ядер, то она будет зависеть от геометрии ядерного остова молекулы. Поэтому электронная волновая функция должна по своим свойствам симметрии относится к одному из типов симметрии той точечной группы, к которой относится равновесная конфигурация рассматриваемой молекулы. Число типов электронных состояний равно числу типов симметрии данной точечной группы. Для абелевых групп это число равно порядку группы, а для других групп равно числу классов.
Классификация электронных состояний по типам симметрии такая же, как и классификация колебательных состояний по типам симметрии (см. гл. 4). Для групп низшей симметрии получаются только невырожденные состояния типа А и В, для группы средней и высшей симметрии будут встречаться наряду с невырожденными состояниями тапа А и В состояния вырожденных типов симметрии E и F. Трижды вырожденные типы симметрии встречаются для групп высшей симметрии, правда, для многоатомных молекул эти группы встречаются довольно редко. Если молекула имеет центр симметрии, то все электронные состояния делятся на четные (типа g) и нечетные (типа u).
В спектроскопии показано, что для нелинейных многоатомных молекул возможны только невырожденные состояния, так как вырожденные для них неустойчивы. Это положение в литературе называют эффектом Яна – Теллера. Оно известно в качестве теоремы Яна – Теллера.
Вырождение, которое мы рассматриваем, есть орбитальное вырождение, связанное с пространственным расположением электронного облака. Наряду с орбитальным вырождением большое значение имеет характеристика электронных состояний, связанная мультиплетностью æ = 2 S + 1, где S значение спинового квантового числа, определяющего полный спиновый момент. В принципе, как и для атомов, для молекул с четным числом электронов S может принимать значения S = 0, 1, 2,…,
а для молекул с нечетным числом электронов S = 1/2, 3/2…
Однако если для молекул реализуется только невырожденные состояния, то по принципу Паули в этом состоянии могут находиться не более двух электронов. В основном электронном состоянии будут, как правило, заполненные электронные оболочки и полный спин (S = 0). При возбуждении молекулы один из электронов переходит в незаполненную оболочку и возникают два неэквивалентных электрона, что приводит к значениям S = 0, 1. Первый случай соответствует антипараллельным, а второй параллельным спинам электронов. Таким образом, основное состояние молекулы будет синглетным (при S = 0, æ = 1) или триплетным (при S = 1, æ = 3).
Для молекул с нечетным числом электронов (как правило, для радикалов) в основном состоянии будет один неспаренный электрон и полный спин равен спину этого электрона S = 1/2, что приводит к дублетному состоянию. Такое значение спина будет осуществляться и для возбужденных состояний, поэтому они будут также дублетными. Число и состояние электронов во внешних электронных оболочках существенно зависит от числа и типа химических связей в многоатомной молекуле.
Рассмотрим кратко химические связи в органических молекулах, основываясь на сведениях о химических связях в двухатомных молекулах. В многоатомной молекуле всегда найдутся атомы, которые связаны с большим числом атомов, чем в двухатомной молекуле. Поэтому возникает вопрос о направленности химических связей в многоатомной молекуле и расположении этих связей. В молекулах с ковалентными связями, по аналогии с двухатомными молекулами, предполагается, что двойные связи осуществляются двумя парами электронов, а тройные – тремя. Деление электронов по типам связи сохраняется в соответствии с проекцией орбитального момента электрона на направление связи (сохраняет смысл понятие о 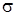 - и p-электронах). Для -связи l = 0, а p-связи l = 1.
- и p-электронах). Для -связи l = 0, а p-связи l = 1.
6.1.2 Химические связи в многоатомных молекулах
Химическая связь в многоатомной молекуле образуется за счет перекрывания электронных облаков s -, p -, d -электронов, аналогично тому, как это было рассмотрено для двухатомной молекулы. Однако в большинстве случаев, начиная от простейших многоатомных молекул (например, молекулы H2O и CH4) и кончая более сложными органическими молекулами, приходится допускать смешивание атомных орбиталей с получением более сложных, так называемых гибридных орбиталей, которые впервые были введены в химию Полингом и Слетером для объяснения строения и свойств многочисленных классов органических соединений. Рассмотрим возможные типы гибридизации и связанное с этими типами строение молекул, а также общую запись молекулярной волновой функции, описывающей распределение электронной плотности.
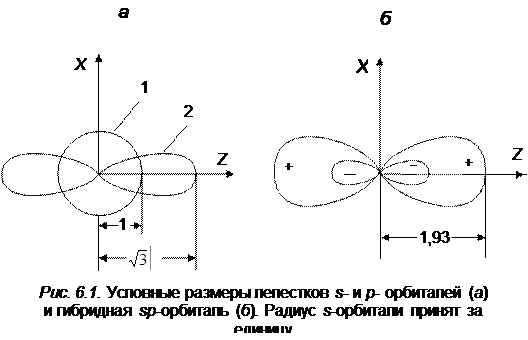
1. Гибридизация sp. При смешивании атомных s- и p -орбиталей одного атома получаются две гибридные орбитали с более вытянутыми лепестками, что способствует более сильному перекрыванию полученных орбиталей с другими атомными орбиталями. условные размеры лепестков орбиталей s и p, а также гибридных sp -орбиталей приведены на рис. 6.1.
Нормированные волновые функции двух гибридных орбиталей запишутся в виде
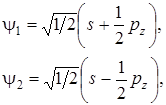 (6.1)
(6.1)
где s – атомная волновая функция s электрона; рz ‑ атомная волновая функция p электрона; sp -гибридизация приводит к линейному строению молекул. (например, молекула BeH2, CO2 и др.)
2. Гибридизация sp2 В результате гибридизации sp2 смешиваются две p -орбитали с одной s -орбиталью. Получаются три гибридные орбитали, лепестки которых расположены в одной плоскости и направлены под углом 120 градусов друг к другу (см. рис 6.2).
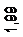
Выберем систему координат так, чтобы одна из координатных осей (Oz) проходила через центральный атом и один из лепестков гибридной орбитали, а ось х перпендикулярно оси z в плоскости чертежа, то волновые функции трех гибридных орбиталей запишутся в следующем виде:
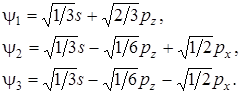 (6.2)
(6.2)
Молекулы, в которых осуществляется sp2 -гибридизация имеют плоское строение, углы между связями равны 120°. К указанным молекулам относятся неорганические ионы типа NO3‑, CO2‑, органические молекулы типа H2CO и др.
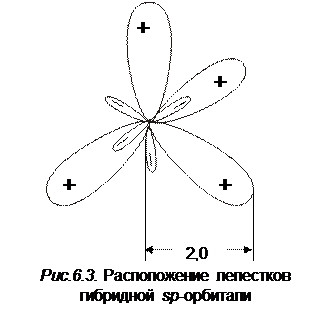
3. гибридизация sp3. Она осуществляется у молекул тетраэдрического строения, например, CH4 и SО42‑. Объединяя атомные орбитали 2s, 2px, 2py, 2pz в молекуле СН4 можно получить четыре эквивалентные орбитали, локализованные на атоме углерода. Такие орбитали носят название гибридных sp 3-орбиталей. Каждая такая гибридная sp 3-орбиталь на ¼ имеет s -характер и на ¾ – p -характер. Лепестки гибридных sp 3-орбиталей приведены на рис 6.3. Строятся они следующим образом. Если атом углерода поместить в центр куба, то максимальную протяженность гибридной sp 3-орбитали нужно проводить к противолежащим вершинам
куба.
Нормированные волновые функции четырех эквивалентных гибридных sp 3- орбиталей имеют вид:
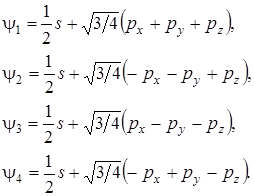 (6.3)
(6.3)
С точки зрения теории МО нет необходимости прибегать к гибридизации орбиталей. Сама запись общей молекулярной функции Y уже будет включать всевозможные комбинации атомных волновых функций, допускающие типы симметрии данной точечной группы, к которой относится рассматриваемая молекула.
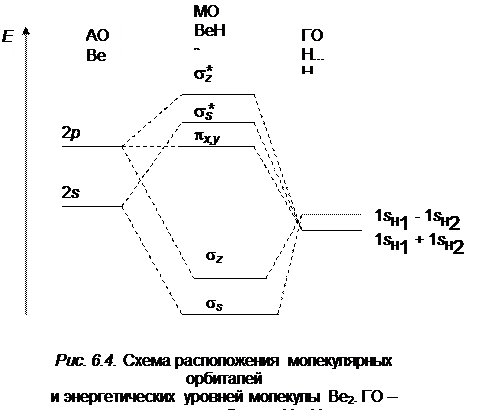
Примеры построения молекулярных орбиталей и соответствующих уровней энергии рассмотрим на ряде простых и более сложных молекул. В линейных молекулах типа АВ2, (например, ВеН2, СО2) сначала составляют групповые орбитали атомов В, которые получаются по тому же способу, что и в двухатомной молекуле В2. После этого определяют принадлежность к s- или p-типу атомных орбиталей центрального атома А. s -орбитали атома всегда образуют s-связи (обычно обозначаются s g). Так как p-орбитали нечетные, то при образовании ими s-связи будем обозначать ее s u. Если перекрываются pz -орбитали с образованием s-связи, то px -и py -орбитали будут образовывать p-связи, обозначаемые p u (иногда p x,y). Только после этого получают МО молекулы АВ2 как линейную комбинацию Ао атома А с групповыми орбиталями атомов В–В.
Рассмотрим молекулярные орбитали линейной трехатомной молекулы ВеН2. Примем ось симметрии молекулы за ось z. Атом Ве имеет валентные 2 s и 2 p орбитали, а атомы водорода – валентные 1 s орбитали. МО орбилаль в молекуле ВеН2 образуется за счет 2 s и 2 pz -орбиталей Ве
и 1 s -орбиталей атомов водорода путем составления их линейных комбинаций. Атомным  - и
- и  -орбиталям приписывается тот же знак (+ или –), который имеет перекрывающаяся с ними часть 2 pz -орбитали. В результате образуются связывающие МО, для которых плотность электронного облака повышена в области между ядрами. Так как 2 s -орбиталь не меняет знака по всей граничной поверхности, то в выражение для связывающей МО с ее участием должна входить сумма
-орбиталям приписывается тот же знак (+ или –), который имеет перекрывающаяся с ними часть 2 pz -орбитали. В результате образуются связывающие МО, для которых плотность электронного облака повышена в области между ядрами. Так как 2 s -орбиталь не меняет знака по всей граничной поверхности, то в выражение для связывающей МО с ее участием должна входить сумма 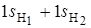 .
.
2 p -орбиталь, на которую переходит один из электронов при образовании связи, имеет две области с противоположными знаками и для образования связывающей МО волновые функции атомов водорода должны быть взяты в виде разности (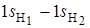 ).
).
Таким образом, получаются две различные связывающие молекулярные орбитали:
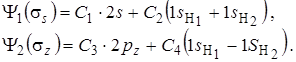 (6.4)
(6.4)
соответствующие им разрыхляющие орбитали будут иметь вид:
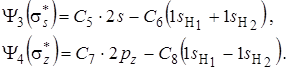 (6. 5)
(6. 5)
Знание коэффициентов С 1, С 2, …, С 8 дает возможность определить электронную плотность на атомах бериллия и водорода. Орбитали 2 px и 2 py в атоме бериллия не принимают участия в образовании связей,
поскольку водород не имеет атомных орбиталей, способных к
p-взаимодействию. Эти орбитали в молекуле BeH2 являются несвязывающими.
Схема энергетических уровней молекулы BeH2 представлена на рис. 6.4. Наиболее глубоко расположенны уровни энергии связывающих орбиталей, затем – несвязывающих, и выше всех располагаются уровни разрыхляющих орбиталей (разрыхляющие орбитали отмечены *). Основное состояние молекулы BeH2 определяется путем заполнения валентными электронами наиболее устойчивых МО. Четыре валентных электрона располагаются на связывающих сигма-орбиталях. Следовательно, электронная конфигурация основного состояния имеет вид:
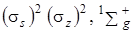 .
.
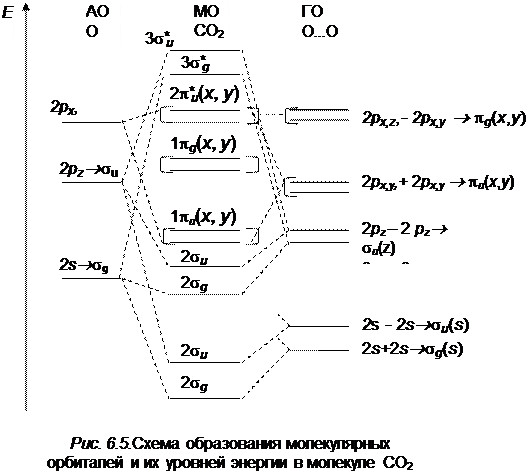
Рассмотрим образование молекулярных орбиталей на примере трехатомной молекулы CO2.
Атом углерода имеет четыре валентных электрона, а кислорода – шесть. Таким образом, нам нужно распределить 16 электронов на соответствующих молекулярных орбиталях. При образовании молекулы СО2 необходимо учесть, что один из s -электронов атома углерода может переходить в p -состояние. Схема молекулярных орбиталей показана на рис. 6.5. Таким образом, 16 электронов молекулы СО2 располагаются на следующих молекулярных орбиталях: (1s g)2 (1s u)2 (2s g)2 (s u)2 (1p u)4 (1p g)4 c образованием основных состояний (1å g). При поглощении света электрон переходит из несвязывающей 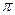 g -орбитали на разрыхляющую p u *-орбиталь.
g -орбитали на разрыхляющую p u *-орбиталь.
6.2. Симметрия молекулярных орбиталей и типы
электронных переходов в нелинейных
многоатомных молекулах
6.2.1. Молекула воды Н2О
Простейшей нелинейной трехатомной молекулой является молекула воды. Она имеет треугольную форму, угол между связями О–Н равен 105. Точечная группа симметрии ‑ С 2u Рассмотрим типы симметрии, по которым преобразуются групповые орбитали атомов водорода и 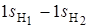 . Если расположить молекулу Н2О так, чтобы ее плоскость совпадала с плоскостью zoy, то групповые орбитали атомов водорода будут преобразовываться по типам симметрии
. Если расположить молекулу Н2О так, чтобы ее плоскость совпадала с плоскостью zoy, то групповые орбитали атомов водорода будут преобразовываться по типам симметрии 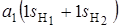 и
и 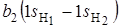 .
.
Атомные орбитали преобразуются следующим образом:
2s ® a 1,
2px ® b 1,
2py® b 2,
2pz® a 1,
Малые буквы латинского алфавита будем использовать для обозначения типов симметрии молекулярных орбиталей.
Схема уровней энергии молекулярных орбиталей молекулы воды приведена на рис. 6.6.
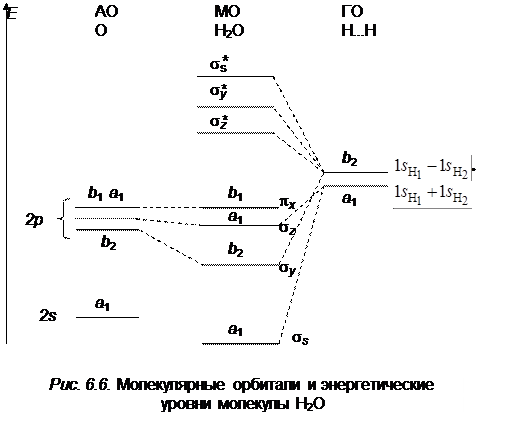
Наиболее сильно связывающими орбиталями являются s s и s u симметрии a 1 и b 2 соответственно. Слабосвязывающей является орбиталь s z симметрии a 1 и несвязывающей является орбиталь p x симметрии b 1. Восемь валентных электронов молекулы воды располагаются на следующих молекулярных орбиталях: (s s)2 (s y)2 (sz y)2 (p x)2 или, используя обозначения по типам симметрии, можем записать:
(a 1)2 (b 2)2 (a 1)2 (b 1)2.
Таким образом, результирующий тип симметрии нижнего электронного состояния будет равен А 1 (он определяется прямым произведением типов симметрии заполненных молекулярных орбиталей).
Первый переход соответствует переходу электрона с 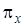 несвязывающей орбитали (b 1) на z * разрыхляющую орбиталь симметрии а 1, т. е. происходит переход с орбитали b 1 на орбиталь а 1. Результирующий переход будет иметь симметрию
несвязывающей орбитали (b 1) на z * разрыхляющую орбиталь симметрии а 1, т. е. происходит переход с орбитали b 1 на орбиталь а 1. Результирующий переход будет иметь симметрию 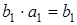 .Таким образом, первый электронный переход в молекуле воды есть переход типа 1А1
.Таким образом, первый электронный переход в молекуле воды есть переход типа 1А1 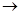 1В1 (результирующие типы электронных состояний обозначаем заглавными буквами латинского алфавита). Спектр, соответствующий этому переходу, расположен в далекой ультрафиолетовой области. Переход в дипольном приближении разрешен по симметрии.
1В1 (результирующие типы электронных состояний обозначаем заглавными буквами латинского алфавита). Спектр, соответствующий этому переходу, расположен в далекой ультрафиолетовой области. Переход в дипольном приближении разрешен по симметрии.
6.2.2. Молекулярные орбитали молекулы формальдегида Н2СО
Химические связи в молекуле формальдегида образуются s - и p - электронами. Так как молекула Н2СО в нижнем электронном состоянии плоская и относится к точечной группе симметрии C 2u, то для атома углерода осуществляется sp 2-гибридизация, которая и приводит к плоскому строению молекулы. Наглядный вид образования связей в молекуле приведен на рис 6.7. В молекуле образуются - и 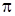 -связи и остаются электроны (n электроны атома кислорода), находящиеся на не связывающих орбиталях. Последовательность молекулярных орбиталей и расположение электронов на них выглядит следующим образом:
-связи и остаются электроны (n электроны атома кислорода), находящиеся на не связывающих орбиталях. Последовательность молекулярных орбиталей и расположение электронов на них выглядит следующим образом:
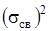
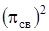
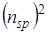
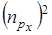 .
.
Разрыхляющие орбитали 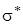 и
и 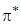 остаются свободными. Таким образом, в группе атомов С = 0 все валентные электроны располагаем на своих орбиталях (для атома кислорода рассматриваем только p -электроны).
остаются свободными. Таким образом, в группе атомов С = 0 все валентные электроны располагаем на своих орбиталях (для атома кислорода рассматриваем только p -электроны).
Почему на несвязывающих орбиталях у нас получилось по два электрона с различной энергией. Дело в том, что один p -электрон атома кислорода смешивается с s -электроном и частично задействован с -связи с атомом углерода. Расчет показывает, что распределение электронной плотности для него обладает шаровой симметрией. В точечной группе симметрии C 2u его орбиталь относится к типу симметрии а 1 и по значению энергии расположена ниже орбитали px -электрона. Симметрия свободной орбитали px относится к типу b 1, который определяется из рассмотрения операций симметрии для группы C 2u. Ось у направлена перпендикулярно плоскости молекулы.
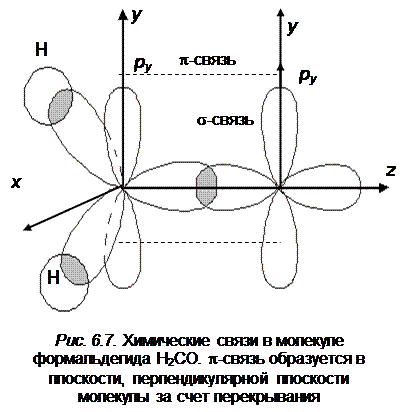
Электронная плотность для орбитали p-типа относится к типу b 2 симметрии. Это проверяется по поведению волновой функции, описывающей распределение pz- электронов, при выполнении операций симметрии. На рис. 6.8. приведена схема расположения уровней энергии валентных электронов молекулы формальдегида и симметрия их молекулярных орбиталей.
Зная симметрию молекулярных орбиталей, можно установить симметрию нижнего электронного состояния как групповое произведение их симметрий молекулярных орбиталей. Итак
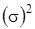
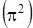
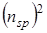
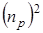
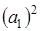
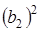
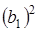 .
.
Произведение типа симметрии самого на себя всегда дает полносимметричное представление (исключая вырожденные состояния), т. е. 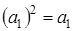 или
или 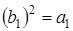 . Таким образом, нижнее состояние молекулы формальдегида относится к полносимметричному представлению А 1. Переход электрона с заполненной несвязывающей p -орбитали (npx) на разрыхляющую p*-орбиталь приводит к спектру поглощения в самой длинноволновой области. На рис 6.8 этот переход обозначается вертикальной стрелкой. Определим симметрию этого перехода. Электрон переходит с молекулярной орбитали симметрии b 1 на разрыхляющую орбиталь p* симметрии b 2. При этом переходе электроны распределяются следующим образом:
. Таким образом, нижнее состояние молекулы формальдегида относится к полносимметричному представлению А 1. Переход электрона с заполненной несвязывающей p -орбитали (npx) на разрыхляющую p*-орбиталь приводит к спектру поглощения в самой длинноволновой области. На рис 6.8 этот переход обозначается вертикальной стрелкой. Определим симметрию этого перехода. Электрон переходит с молекулярной орбитали симметрии b 1 на разрыхляющую орбиталь p* симметрии b 2. При этом переходе электроны распределяются следующим образом:
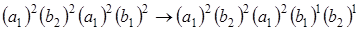 .
.
Симметрия электронного состояния (p*) будет определяться прямым произведением симметрий переходов, или в конечном итоге будет определяться произведением 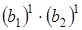 .
.

Если произвести перемножение типа симметрии b1 на тип симметрии b 2(см. таблицу характеров для точечной группы симметрии C 2u), то мы получим тип симметрии а2. с точки зрения типов симметрии электронных состояний этот переход обозначаем как A 1® A 2.
Анализируя таблицу характеров для этого типа перехода, мы видим, что он запрещен в дипольном приближении.
Рассуждая аналогичным образом, найдем тип симметрии перехода 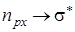 . Этот переход типа А 1® В 1, который в дипольном приближении разрешен.
. Этот переход типа А 1® В 1, который в дипольном приближении разрешен.
Таким образом, интерпретируя спектр поглощения формальдегида, который в ближнем ультрафиолете состоит из двух полос 270 нм (слабая) и 185 нм (сильная), можем резюмировать, что первый относится к типу 1 A 1®1 A 2 (запрещен в дипольном приближении), а второй - к типу симметрии 1 A 1®1 B 1 (разрешен в дипольном приближении с силой осциллятора, близкой к единице). Цифра  слева вверху означает синглетность перехода, т. е. переходы совершаются без изменения спинового числа (
слева вверху означает синглетность перехода, т. е. переходы совершаются без изменения спинового числа (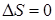 ).
).
Слабое проявление поглощения в области 270 нм может быть связано с вибронным заимствованием интенсивности (см. спектр поглощения бензола). Можно показать, что за счет вибронного взаимодействия колебание b 1 в молекуле формальдегида (колебания в молекуле Н2СО распределяется по следующим типам симметрии: 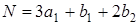 ) может быть ответственно за разрешение первого электронного перехода, так какпрямое произведение А2 на b 1 дает тип симметрии B2, который разрешен по правилам отбора. Таким образом, за счет вибронного взаимодействия с участием колебания b1 первый электронный переход в молекуле формальдегида Н2СО частично разрешается.
) может быть ответственно за разрешение первого электронного перехода, так какпрямое произведение А2 на b 1 дает тип симметрии B2, который разрешен по правилам отбора. Таким образом, за счет вибронного взаимодействия с участием колебания b1 первый электронный переход в молекуле формальдегида Н2СО частично разрешается.
6.2.3. молекула бензола С6Н6
В молекуле бензола 30 валентных электронов ‑ 2 s - и 2 p -электронов атомов углерода (6×4 = 24) и шесть 1 s электронов атомов водорода - образуют химические связи. Каждый атом углерода соединен с двумя соседними атомами углерода и одним водородом. При образовании этой молекулы осуществляется для атома углерода sp2 -гибридизация. При этом в атоме углерода задействованы два 2 р и один 2 s электрон. Один электрон атома углерода своей 2 рz -орбиталью ориентирован перпендикулярно плоскости и не задействован в s-связях гибридной sp 2-орбитали. Гибридная орбиталь в плоскости молекулы образует три s-связи: две s-связи с атомами углерода и одну s-связь с атомом водорода. В этих связях задействовано 24 электрона (см. рис. 6.9).
Расположены эти s-связи под углом 120° в одной плоскости в полном соответствии с рентгеноструктурными данными. Этим объясняется плоское строение молекулы бензола, имеющей форму правильного шестиугольника. Орбитали негибридизированных шести рz -электронов кольца молекулы расположены перпендикулярно плоскости (см. рис. 6.10).
На рис. 6.10 изображены граничные поверхности волновых функций р -электронов атомов углерода. Прямые отрезки линий указывают на возможность перекрытия электронных облаков р -электронов.
Каждая р -орбиталь перекрывается с другими р - орбиталями с двух сторон. Поскольку в молекуле бензола все р -орбитали перекрываются друг с другом, то каждый р -электрон может быть около любого из атомов углерода. Эти электроны образуют p-связи в молекуле бензола. При этом невозможно указать, каким атомам углерода принадлежит каждая из трех пар электронов, образующих эти связи. Поэтому говорят, что в молекуле бензола p-связи являются делокализованными, т. е.
p-электроны в молекуле бензола свободно передвигаются в плоскости кольца углеродных атомов. Для полного насыщения p-связей в молекуле С6Н6 необходимо 12 р -электронов, а в действительности имеется только шесть, т. е. p-связи заполнены электронами только наполовину. Поскольку p- связи в молекуле бензола являются делокализованными, то структурную формулу C6H6 изображают следующим образом: 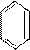 .
.

Отрезками прямых показаны делокализованные p-связи. О том, что связи в молекуле бензола по своему характеру являются промежуточными между одинарными и двойными свидетельствует тот факт, что их длина, равная 1,40 Å, лежит между длинами одинарной и двойной связей, равных соответственно 1,54 и 1,32 Å (с точностью до 0,02 Ǻ).
Молекула бензола имеет плоское строение и относится к точечной группе симметрии D 6 h (см. рис. 4.7 гл. 4). Осью высшего порядка является ось симметрии шестого порядка, проходящая перпендикулярно плоскости рисунка. Кроме того, вдоль оси шестого порядка расположены две оси 3-го и одна ось 2-го порядка. В плоскости молекулы, перпендикулярной оси шестого порядка, расположены две группы осей второго порядка, каждая из которых состоит из трех элементов (обозначены оси соответственно  и
и  ). Молекула имеет горизонтальную плоскость симметрии s h (совпадающую с плоскостью ху), три вертикальные плоскости su и три диагональные плоскости sd, одна из которых совпадает с плос
). Молекула имеет горизонтальную плоскость симметрии s h (совпадающую с плоскостью ху), три вертикальные плоскости su и три диагональные плоскости sd, одна из которых совпадает с плос  костью yz. Центр инверсии i совпадает с началом координат. Кроме перечисленных элементов симметрии молекула имеет по две зеркально-поворотные оси типа S 3 и S 6. В итоге получаем точечную группу симметрии D6 h, которая включает следующие операции симметрии: I,2 C 6, 2 C 3, C 2, 3 С 2' ,3C 2¢¢, i, 2 S 3, 2 S 6, s h, 3s d, 3su.
костью yz. Центр инверсии i совпадает с началом координат. Кроме перечисленных элементов симметрии молекула имеет по две зеркально-поворотные оси типа S 3 и S 6. В итоге получаем точечную группу симметрии D6 h, которая включает следующие операции симметрии: I,2 C 6, 2 C 3, C 2, 3 С 2' ,3C 2¢¢, i, 2 S 3, 2 S 6, s h, 3s d, 3su.
Для отнесения электронных состояний молекулы бензола по типам симметрии воспользуемся методом молекулярных орбиталей (МО), который позволяет находить результирующую симметрию электронного состояния на основании данных о симметрии волновых функций, описывающих это состояние. Будем рассматривать только самые внешние шесть 2 р электронов. Для них запишем молекулярную орбиталь в виде
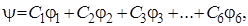 (6.6)
(6.6)
где j1, j2,…j6 – атомные pz – орбитали атомов углерода. Коэффициенты Сi находятся из условия 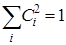 (i = 1, 2, …, 6). Общий вид волновых функций (МО) по Хюккелю можно записать следующим образом:
(i = 1, 2, …, 6). Общий вид волновых функций (МО) по Хюккелю можно записать следующим образом:
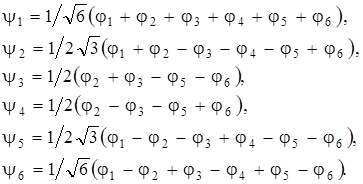 (6.7)
(6.7)
Решая уравнение Шредингера с указанными волновыми функциями, получим шесть значений энергии Е 1, Е 2 ,...Е 6, соответствующих шести МО. Однако, значения энергии Е 2= Е 3, и Е 4= Е 5равны, т. е. волновые функции  2 и 3, а также 4 и 5 составляют вырожденные наборы орбиталей. Для установления симметрии этих орбиталей воспользуемся теорией групп.
2 и 3, а также 4 и 5 составляют вырожденные наборы орбиталей. Для установления симметрии этих орбиталей воспользуемся теорией групп.
Атомная орбиталь j i атома углерода (2 pz -орбиталь) антисимметрична по отношению к плоскости молекулы. Свойства преобразований шести функций j i под действием различных элементов симметрии точечной группы D 6 h можно получить следующим образом. Например, рассмотрим операцию поворота молекулы С6Н6 на 60° вокруг оси шестого порядка. При этой операции функция j1 переходит в j2, а функция j2 в j3 и т.д. Характер этой операции симметрии будет равен нулю, так как при этой операции ни один из атомов не остается на месте. Операция отражения в плоскости молекулы (s h) переводит каждую функцию в саму себя с изменением знака на обратный. Легко показать, что характер этой операции равен 6. Операция отражения в плоскости симметрии, проходящей через атомы С1 и С4, оставляет j1 и j4 без изменений, а функцию j2 переводит в j6, а j3 в j5. Характер этой операции будет равен +2. Аналогичным образом можно рассмотреть и все остальные операции симметрии. Мы получаем таким образом приводимое представление точечной группы симметрии D6h, которое будет иметь следующие характеры:
| I | C 6 | C 3 | C 2 | C 2 ' | C 2 '' | i | S3 | S6 | s h | s d | su | |
| Г (R) | +6 | ‑2 | ‑6 | +2 |
Этот набор чисел складывается из наборов характеров неприводимых представлений a 2 u , b 2 g , e 1 g , e 2 u . Следовательно, орбитальные волновые функции, получающиеся из 2 p z – атомных функций углерода, всегда должны принадлежать указанным выше неприводимым представлениям, если симметрия молекулы сохраняется D 6 h.
Первые три орбитали 1, 2 и 3 будут связывающими, и по энергии они расположены ниже, чем остальные три. Первая орбиталь относится к типу симметрии a 2 u, 2 и 3составляют вырожденный набор орбиталей типа симметрии e 1 g с одним узлом в плоскости молекулы. Орбитали 4 и Y5 составляют вырожденный набор разрыхляюших орбиталей с двумя узлами в плоскости молекулы (тип симметрии e 2 u). 6 – орбиталь характеризуется разрыхляющим характером и содержит три узла в плоскости молекулы (тип симметрии b 2 g).
Схематически вид орбиталей показан на рис. 6.11. Здесь приведена только часть волновых функций, которая расположена над плоскостью (фаза атомной орбитали на i -м атоме углерода определяется знаком функции j i в разложениях).

Расстояния от контурных линий, которыми обведены атомы, до каждого из них пропорциональны величине коэффициента при орбитали разложения.
Сплошными линиями обозначены зоны положительных значений молекулярных орбиталей, а штриховыми – зоны отрицательных значений. Узловые плоскости, проходящие через ось z, отмечены штриховыми линиями. Приведенные функции имеют соответсвенно 0, 1, 2, и 3 узловые плоскости. Энергия орбиталей растет с увеличением числа узловых плоскостей. Наименьшей энергией обладает орбиталь симметрии a2u, не имеющая узловых плоскостей. Затем в порядке возрастания энергии следуют e 1 g и e 2 u – орбитали. Наконец, самой высокой оказывается орбиталь b 2 g с тремя узлами в плоскости молекулы.
Общая схема энергетических уровней молекулы бензола приведена на рис.6.12
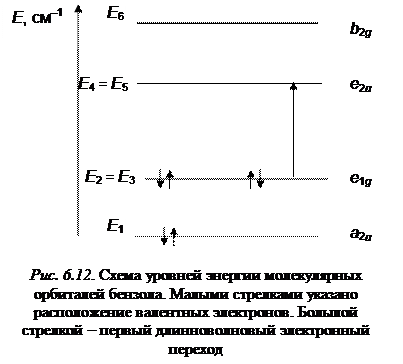
Все шесть p-электронов молекулы бензола согласно принципу Паули займут два нижних энергетических состояния: 2 электрона на орбитали a 2 u и 4 электрона на орбитали e 1 g. Основная конфигурация бензола (учитывается только его p-электронная система) записывается следующим образом:
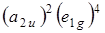 .
.
С точки зрения теории групп заполненные подоболочки всегда относятся к полносимметричному представлению. Таким образом, нижнее состояние молекулы бензола A1 g. При поглощении кванта света электрон из молекулярной орбитали e 1 g переводится на орбиталь e2u C точки зрения электронной конфигурации это записывается следующим образом
(a 2 u)2(e 1 g)4 ®(a 2 u)2(e 1 g)3 (e 2 u)1.
Для определения симметрии этого состояния надо найти прямое произведение скобок. Однако ясно, что
a 2 u ×a 2 u = a 1 g , а e 1 g × e 1 g ×e 1 g = e 1 g .
следовательно, симметрия результирующего состояниия (a 2 u )2(e 1 g )3(e 2 u )1 есть прямое произведение e 1 g × e 2 u = B 2 u + B 1 u + E 1 u . Симметрия электронных состояний обозначается большими прописными буквами.
Таким образом, первое возбужденное состояние бензола дает три отрбитальных состояния, которые соответствуют случайному вырождению. За счет электронного взаимодействия вырождение снимается, и эти состояния располагаются в порядке возрастания энергии B 2 u , B 1 u и E 1 u . Расположение указанных состояний по энергии производится путем сравнения результатов теоретического расчета с экспериментом. Следовательно, первый длинноволнывой электронный переход типа 1 А 1 g ®1 B 2 u в молекуле бензола запрещен по симметрии (см. рис. 6.27).
6.3. Принцип Франка – Кондона
для многоатомных молекул
Прежде чем рассматривать электронные спектры конкретных молекул, предварительно познакомимся с одним из важнейших условий, формирующих электронно-колебательные спектры многоатомных молекул, принципом Франка – Кондона. Он впервые был сформулирован Франком и обоснован в квантовой механике Кондоном и касался электронно-колебательного спектра двухатомной молекулы. Основывался он на адиабатическом приближении, т. е. предполагалось, что в двухатомной молекуле имеются частицы (ядра), которые движутся сравнительно медленно по отношению к электронам. Поэтому во многих задачах механики можно рассматривать движение электронов в поле неподвижных ядер. Формулировка этого принципа выглядела следующим образом: при поглощении света переход электрона из нижнего электронного состояния в верхнее (возбужденное) происходит настолько быстро, что за это время ни скорость ядер, ни положение их в пространстве заметно не изменяется. Применение этого принципа к многоатомным молекулам позволяет понять многие черты формирования электронно-колебательного спектра многоатомной молекулы.
Особенность многоатомной нелинейной молекулы, по сравнению с двухатомной состоит в том, что многоатомная молекула имеет 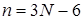 колебательных степеней свободы в противоположность одной колебательной степени свободы для двухатомной молекулы. Для рассмотрения электронно-колебательных переходов в двухатомной молекуле мы брали потенциальные кривые нижнего и верхнего электронных состояний на плоскости и проводили вертикальные стрелки, изображающие электронные переходы.
колебательных степеней свободы в противоположность одной колебательной степени свободы для двухатомной молекулы. Для рассмотрения электронно-колебательных переходов в двухатомной молекуле мы брали потенциальные кривые нижнего и верхнего электронных состояний на плоскости и проводили вертикальные стрелки, изображающие электронные переходы.
В случае многоатомной молекулы форма потенциальной поверхности значительно сложнее. Она имеет большое число различных минимумов, глубина которых может быть весьма различной. Под минимумами поверхности будем понимать такие ее точки Q 1, Q 2, … Qn, в которых 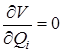 для всех координат, характеризующих расстояния ядер друг относительно друга. Эти точки соответствуют положениям равновесия молекул, которые не могут быть нарушены без воздействия какого-то внешнего фактора.
для всех координат, характеризующих расстояния ядер друг относительно друга. Эти точки соответствуют положениям равновесия молекул, которые не могут быть нарушены без воздействия какого-то внешнего фактора.
Тем не менее иногда пытаются представить потенциальную поверхность геометрически в виде некоторой поверхности в пространстве n + 1 измерений. Такое представление позволяет использовать по аналогии выводы, полученные для двухатомных молекул. Для этого произведем сечение многомерной поверхности по одной из нормальных координат, скажем, Qi. тогда на плоскости мы можем рассматривать профиль сечения, некую одномерную потенциальную поверхность, зависящую от одной Qi координаты. Мы, таким образом, пришли к полной аналогии с двухатомной молекулой, в которой потенциальная поверхность рассматривается как функция относительного расстояния между атомами.
Таким образом, наиболее существенные результаты применения принципа Франка – Кодона к многоатомным молекулам можно иллюстрировать при помощи двухмерной диаграммы, а при рассмотрении зависимости потенциальной энергии от одной из нормальных координат Qi можно применять диаграммы 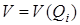 , аналогичные диаграммам
, аналогичные диаграммам 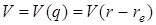 для двухатомных молекул.
для двухатомных молекул.
Формулировка принципа Франка – Кондона для многоатомной молекулы сохраняется такая же как и для двухатомной только добавляется сохранение мгновенной конфигурации ядер, т. е. сохранение ее симметрии. Действительно, если симметрия молекулы не изменяется в процессе электронного возбуждения, то легко видеть, что мгновенная конфигурация ядер, возникшая в результате возбуждения и идентичная равновесной конфигурации ядер основного состояния, должна сохраняться. Это имеет место при переходах из нулевого колебательного состояния, в котором находится большая часть молекул при сравнительно низких температурах.
Для многоатомной молекулы существуют колебания полносимметричного и неполносимметричного типа, и они по-разному будут проявляться в электронном спектре. Так как колебания полносимметричного типа не изменяют симметрию равновесного состояния молекул, то они в спектре могут проявляться несколькими квантами при любом сдвиге кривых потенциальной энергии по нормальной координате 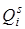 .
.
Начальная конфигурация молекулы будет равновесной, если будут возбуждены только полносимметричные колебания. Плотность вероятности пребывания системы в данном состоянии при 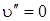 наибольшая при равновесном состоянии между ядрами. На рис. 6.13 стрелкой указан наиболее интенсивный переход
наибольшая при равновесном состоянии между ядрами. На рис. 6.13 стрелкой указан наиболее интенсивный переход 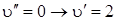 из нижнего электронного состояния в верхнее для симметричной координаты .
из нижнего электронного состояния в верхнее для симметричной координаты .
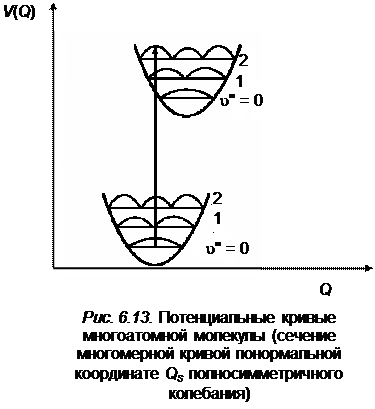
как видно из рисунка, положение потенциальной кривой верхнего состояния может быть сдвинуто относительно нижнего состояния на любую величину 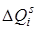 без изменения симметрии равновесной конфигурации. Поэтому полносимметричные колебания могут проявляться несколькими квантами с различной интенсивностью в полной аналогии с двухатомной молекулой, в которой имеется только одно колебание (с точки зрения симметрии оно полносимметрично). Наряду с переходами, в которых возбуждены полносимметричные колебания, возможны также и переходы между состояниями, в которых возбуждены неполносимметричные колебания. Для неполносимметричных колебаний симметрия равновесной конфигурации молекулы сохраняется в момент прохождения атомами положения равновесия, и поэтому на диаграмме потенциальных кривых (рис.6.14) минимумы нижней и верхней потенциальной кривой совпадают, подобно тому, как это имеет место для двуатомной молекулы при
без изменения симметрии равновесной конфигурации. Поэтому полносимметричные колебания могут проявляться несколькими квантами с различной интенсивностью в полной аналогии с двухатомной молекулой, в которой имеется только одно колебание (с точки зрения симметрии оно полносимметрично). Наряду с переходами, в которых возбуждены полносимметричные колебания, возможны также и переходы между состояниями, в которых возбуждены неполносимметричные колебания. Для неполносимметричных колебаний симметрия равновесной конфигурации молекулы сохраняется в момент прохождения атомами положения равновесия, и поэтому на диаграмме потенциальных кривых (рис.6.14) минимумы нижней и верхней потенциальной кривой совпадают, подобно тому, как это имеет место для двуатомной молекулы при 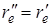 .
.
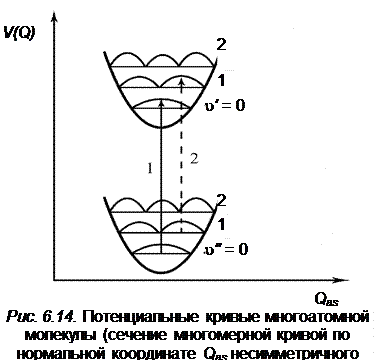
При одинаковой или близкой форме потенциальных поверхностей наиболее вероятными будут переходы 0–0, 1–1 и т. д., как это показано на рис. 6.14 стрелками 1 и 2. Стрелка 2 указана штрихами, так как интенсивность перехода 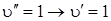 будет весьма мала в силу незначительной заселенности уровня
будет весьма мала в силу незначительной заселенности уровня 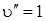 . переход
. переход 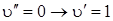 будет слабым ввиду недостаточного перекрытия волновых функций нижнего и верхнего электронных состояний. Если указанный случай для двухатомных молекул не является типичным, то для многоатомных молекул он является основным наряду со случаем сдвинутых минимумов потенциальной энергии для полносимметричных колебаний.
будет слабым ввиду недостаточного перекрытия волновых функций нижнего и верхнего электронных состояний. Если указанный случай для двухатомных молекул не является типичным, то для многоатомных молекул он является основным наряду со случаем сдвинутых минимумов потенциальной энергии для полносимметричных колебаний.
Если учитывать населенность колебательных уровней (при сравнительно низких температурах заселен в основном нулевой колебательный уровень), то наиболее вероятным будет бесколебательный переход 0‑0 и очень слабыми будут последующие переходы для неполносимметричного колебания. Отсюда становится понятным вопрос, почему колебания несимметричного типа проявляются, как правило, одним квантом в электронно-колебательном спектре, а колебания полносимметричного типа могут проявляться многими квантами, давая наблюдаемые повторы (секвенции) полос в спектрах по полносимметричному колебанию (см. спектры поглощения бензола и др.). Что касается полносимметричных колебаний, то их активность в электронном спектре и число наблюдаемых электронно-колебательных полос определяется взаимным расположением потенциальных кривых основного и возбужденного состояний. Для одной и той же многоатомной молекулы число наблюдаемых полос различно для разных типов полносимметричных колебаний, что связано с неодинаковым возмущением электронной оболочки молекул при данных колебаниях. Одни полносимметричные колебания могут давать небольшое число слабых колебательных полос в спектре, а другие – большую последовательность полос в зависимости от величины сдвига минимума потенциальной кривой по полносимметричной координате .
Изложенные выше положения можно получить из квантовомеханического рассмотрения. Вероятность дипольного перехода пропорциональна квадрату матричного элемента M ik дипольного момента перехода, который выражается через волновые функции двух комбинирующих состояний i и k следующим образом:
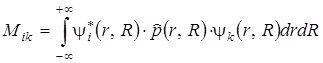 , (6.8)
, (6.8)
где 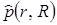 – оператор дипольного момента перехода, dr и dR – элементы объема электронных и ядерных координат
– оператор дипольного момента перехода, dr и dR – элементы объема электронных и ядерных координат 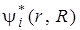 и
и 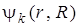 – волновые функции комбинирующих состояний.
– волновые функции комбинирующих состояний.
Выражение (6.8) можно использовать как для чисто электронного перехода, если под 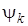 и
и 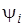 подразумевать электронные волновые функции, так и для электронно-колебательных (вибронных) переходов, если в качестве и и взять вибронные волновые функции, т. е.
подразумевать электронные волновые функции, так и для электронно-колебательных (вибронных) переходов, если в качестве и и взять вибронные волновые функции, т. е.
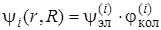 , (6.9)
, (6.9)
Имея в виду, что полный дипольный момент молекулы зависит от движения электронов и ядер, то можно записать, что
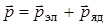 . (6.10)
. (6.10)
Подставим значения волновых функций(6.9) и дипольного момента (6.10) в выражение (6.8), тогда для M ik получим следующее выражение:
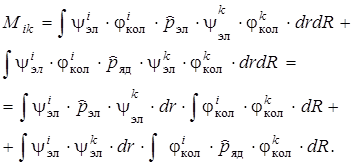 (6.11)
(6.11)
Функции 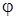 и
и 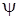 для простоты берутся действительными. Вследствие ортогональности волновых функций различных электронных состояний получим
для простоты берутся действительными. Вследствие ортогональности волновых функций различных электронных состояний получим
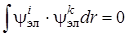 , (6.12)
, (6.12)
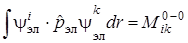 , (6.13)
, (6.13)
где 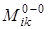 -матричный элемент чисто электронного перехода. После упрощений выражение (6.11) можно окончательно записать следующим образом:
-матричный элемент чисто электронного перехода. После упрощений выражение (6.11) можно окончательно записать следующим образом:
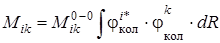 . (6.14)
. (6.14)
Соотношение (6.14) является квантовомеханическим выражением принципа Франка – Кондона для многоатомных молекул и показывает, что вероятность вибронного перехода будет тем больше, чем сильнее наложение колебательных волновых функций комбинирующих электронных состояний. Интеграл в (6.14) называется интегралом наложения колебательных функций в i -м и k -м электронных состояниях. Этот интеграл характеризует распределение интенсивности в спектре между разными колебательными состояниями внутри одноэлектронного перехода (при поглощении света из основного электронного состояния (i = 0)на первое возбужденное состояние(k = 1)). Для того чтобы интеграл Кондона не был равен нулю при изменении колебательного квантового числа u, необходимо, чтобы волновые функции 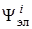 и
и 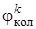 были решениями разных уравнений. Это условие выполняется, если при электронном возбуждении смещаются положения равновесия ядер по данной координате.
были решениями разных уравнений. Это условие выполняется, если при электронном возбуждении смещаются положения равновесия ядер по данной координате.
Приближение Кондона удовлетворительно описывает большое число вибронных переходов с участием полносимметричных колебаний в спектрах с разрешенным чисто электронным переходом. В этом случае матричный элемент дипольного момента электронного перехода определяется при равновесном положении ядер.
Выражение (6.14) и есть квантовомеханическая формулировка принципа Франка – Кондона. Оно приводит к тому же качественному результату, что и полуклассический принцип Франка. Наиболее вероятны переходы, изображаемые на схемах прямыми стрелками. Кроме того, выражение (6.14) позволяет правильно количественно описывать распределение интенсивностей в вибронном спектре. При этом должно выполняться приближение Кондона, т. е. предполагается, что взаимодействие электронного движения с колебательным незначительно. Это необходимо, чтобы можно было представить каждую вибронную волновую функцию в виде произведения 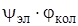 , т. е. должно выполняться приближение Борна-Оппенгеймера. Затем следует предположение, что дипольный момент чисто электронного перехода не зависит от колебательных координат, а взаимодействие нормальных колебаний в обоих электронных состояниях между собой незначительно.
, т. е. должно выполняться приближение Борна-Оппенгеймера. Затем следует предположение, что дипольный момент чисто электронного перехода не зависит от колебательных координат, а взаимодействие нормальных колебаний в обоих электронных состояниях между собой незначительно.
Отметим ряд частных случаев, следующих из принципа Франка – Кондона. Если потенциальные кривые не смещаются и не деформируются при электронном возбуждении, то колебательные волновые функции нижнего и верхнего электронных состояний идентичны. В силу ортонормированности волновых функций разрешены будут только переходы, при которых 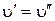 , т. е. интеграл вида (6.14) будет отличен от нуля. При достаточно низких температурах, когда молекулы будут находиться на нулевом колебательном уровне, в спектре будут наблюдаться только 0-0 переходы. При незначительном сдвиге потенциальных поверхностей будет наблюдаться довольно интенсивный 0–0 переход и слабые колебательные спутники. При этом в электронно-колебательном спектре должны быть активны только полносимметричные колебания.
, т. е. интеграл вида (6.14) будет отличен от нуля. При достаточно низких температурах, когда молекулы будут находиться на нулевом колебательном уровне, в спектре будут наблюдаться только 0-0 переходы. При незначительном сдвиге потенциальных поверхностей будет наблюдаться довольно интенсивный 0–0 переход и слабые колебательные спутники. При этом в электронно-колебательном спектре должны быть активны только полносимметричные колебания.
Отметим, что правила отбора, выведенные из рассмотрения свойств интеграла могут нарушаться, если предположение о независимости интеграла от колебательных координат не выполняется. Учет этой зависимости может быть весьма существенным особенно, для больших запасов колебательной энергии, а также в силу малости электронного момента перехода или его запрещенности по симметрии. Предположение о разделении полной электронно-колебательной функции на сомножители, зависящие от электронных и ядерных координат, справедливо лишь при малых запасах колебательной энергии, когда справедливо гармоническое приближение. При больших запасах колебательной энергии взаимодействие различных колебаний может быть настолько велико, что произвести разделение ядерных и электронных координат невозможно.
Мы рассмотрели переходы между электронными состояниями, для которых симметрия равновесной конфигурации одинакова. Однако возможны случаи, когда при возбуждении она изменяется. В случае понижения симметрии одного из комбинирующих состояний может увеличиться число возможных переходов, особенно для вырожденных состояний, когда эти состояния расщепляются (в силу их неустойчивости) и получаются равновесные конфигурации более низкой симметрии. Известно, что чем ниже симметрия молекулы, тем более богат колебаниями ее электронно-колебательный спектр.
6.4. Некоторые вопросы теории
электронно - колебательных спектров
многоатомных молекул