 2017-12-16
2017-12-16 3332
3332Варианты:
1. Кислота титруется ступенчато (H3PO4,H2SO3и др.)
2. Кислота титруется сразу по всем ступеням (H2С2O4,H2SO4и др.)
1) Условие наличия скачка на кривой титрования:
Кд >10-7– 10-8(pK < 7-8)
Количество скачков:
· Если К >10-7 – 10-8Þскачок есть
· Если К < 10-7– 10-8Þскачка нет
2) Условие разделения скачков:  (DрК³4)
(DрК³4)
· Если  ÞIиIIт. э. располагаются на кривой раздельно, кислота титруется ступенчато
ÞIиIIт. э. располагаются на кривой раздельно, кислота титруется ступенчато
·  Если
Если  Þкислота титруется сразу по двум ступеням
Þкислота титруется сразу по двум ступеням
Например, H3РO4: К1= 7,1·10-3; К2= 6,2·10-8; К3= 5,0·10-13
Выбирают индикатор с помощью кривой титрования. Для этого на график кривой титрования наносят интервал перехода индикатора. У правильно подобранного индикатора интервал перехода полностью или частично перекрывается скачком титрования.
Погрешности, обусловленные несовпадением точки эквивалентности и рТ индикатора, называются индикаторными.
Индикаторные погрешности в кислотно-основном титровании удобно разделить на 4 вида:

Водородная индикаторная погрешность может возникнуть при недотитровании сильной кислоты (в таком случае погрешность отрицательная) либо когда сильная кислота используется в качестве титранта и добавлена в избытке (положительная погрешность).
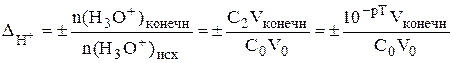
Если концентрации титруемого вещества и титранта одинаковы, то Vконечн = 2V0, тогда

Гидроксидная погрешность может возникнуть при недотитровании сильного основания (отрицательная погрешность) либо в том случае, когда сильное основание используется в качестве титранта и добавлено в избытке (положительная погрешность).

Кислотная и основная индикаторные погрешности могут быть только отрицательными (если, конечно, исключить гипотетический случай использования слабой кислоты или основания в качестве титранта).
Величина кислотной погрешности представляет собой молярную долю неоттитрованной кислоты.


Если  , то
, то 
Формула для расчёта основной погрешности выводится аналогичным образом и выглядит следующим образом


3) Электрохимические методы анализа — группа методов количественного химического анализа, основанные на использовании электролиза.Различают методы, основанные на электродной электрохимической реакции (потенциометрия, полярография, вольтамперометрия, амперометрия, хронопотен-циометрия, электролиз, кулонометрия и др .); методы, не связанные электродной электрохимической реакцией (кондуктометрия, диэлкометрия), и методы, связанные с изменениями структуры двойного электрического слоя (тензометрия). При высокочастотном кондуктометрическом титровании используют токи высокой частоты — от десятков тысяч герц до сотен мегагерц. Отсутствие непосредственного контакта исследуемого раствора с электродами.Данный метод основан на регистрации изменения так называемой высокочастотной электрической проводимости G в процессе титрования. Величина G в свою очередь есть сложная функция от удельной электрической проводимости χ и частоты тока. Для осуществления метода высокочастотного титрования исследуемый раствор подвергается действию высокочастотного электромагнитного поля, создаваемого внутри измерительных ячеек, которые представляют собой либо электрический конденсатор, либо катушку индуктивности. По этому признаку измерительные ячейки делят на: 1) емкостные и 2) индуктивные. Простейшая измерительная ячейка емкостного типа представляет тонкостенный стеклянный сосуд, на наружной поверхности которого монтируются две изолированные друг от друга металлические обкладки, подключенные к высокочастотному генератору. Измерительная ячейка емкостного типа представляет собой стеклянный цилиндрический сосуд, помещаемый между пластинами конденсатора. Последние имеют форму колец.
Билет
1)Комплексные соединения – вещества сложного состава, состоящие из более простых (ионов, молекул), способные к самостоятельному существованию в растворах. В состав комплексного иона или молекулы входит центральный ион – комплексообразователь, вокруг которого координируются ионы или молекулы, которые называются лигандами (аддендами).Реакция образования комплексных соединений в растворах в общем виде описывается уравнением: mM + nL =MmLn, где М – ион металла, L - лиганд. Заряды ионов с целью упрощения не указаны. Константа равновесия для этой реакции равна:  , где [MmLn] – равновесная концентрация комплексного соединения, [M] и [L] – равновесные концентрации иона металла и лиганда соответственно. Данная константа равновесия называется константой образования или константой устойчивости комплексного соединения. Величина, обратная константе устойчивости, называется константой нестойкости данного соединения (Кн):
, где [MmLn] – равновесная концентрация комплексного соединения, [M] и [L] – равновесные концентрации иона металла и лиганда соответственно. Данная константа равновесия называется константой образования или константой устойчивости комплексного соединения. Величина, обратная константе устойчивости, называется константой нестойкости данного соединения (Кн):
Кn =  .
.
Константы устойчивости и нестойкости комплексных соединений характеризуют их прочность. Чем больше значение константы устойчивости (чем меньше значение константы нестойкости), тем больше концентрация комплексного соединения в растворе, и наоборот. Равновесия в растворах комплексных соединений являются равновесиями малодиссоциирующих соединений.
Типы комплекссных соединенеий, используемых в аналитической химии.
1)Комплексы ионов металлов с монодентатными лигандами. (Cl−, F−, CN−, OH−, молекулы NH3, H2O, CO и др. таким образом, в качестве монодентатных лигандов чаще всего выступают неорганические ионы или молекулы). Примерами монодентатных лигандов органической природы могут служить ацетат-ионы (сн3соо−) и анионы других монокарбоновых кислот.
2)Комплексы ионов металлов с полидентатными лигандами. (органические соединения, называемые в химической литературе органическими реагентами).такие лиганды должны содержать не менее двух неподеленных пар электронов, которые будут обеспечивать возможность образования хелатов с ионами метал-лов. поэтому большинство органических реагентов являются хелатообразующими.
3)Катионные хелаты. наиболее распространенными хелатами этой группы являются комплексы полиаминов с ионами металлов.
4)Анионные хелаты. типичными анионными хелатами являются комплексы ионов металлов с этилендиаминотетрауксусной кислотой (ЭДТА).
5)Нейтральные хелаты (внутрикомплексные соединения). При образовании хелата положительный заряд центрального атома нейтрализуется присоединением равного числа отрицательно заряженных лигандов с образованием «внутренней соли».
6)Эфирные хелаты. при образовании хелата аквалиганды гидратированного иона металла вытесняются донорными атомами хелатного лиганда.
Активной называют депротонированную форму лиганда,способную к координации, которая зависитот константыдиссициациилиганда и рНраствора.Условная константа устойчивости.
Константу β, выраженную с учетом активной доли лиганда, называют условной константой.
M + nL = MLn
β = [MLn]/[M]·[L]n из α=[L]/CL; [L]=CLα
β1 = [MLn]/[M]·[CL]n · αn
Условную константу используют в практических расчетах, т.к. она позволяет учитывать активную долю лиганда.
2) рН раствора соли слабого основания и сильной кислоты.
В водном растворе соль ВА практически полностью диссоциирует по уравнению: ВА → В+ + А-Катионы соли (В+) взаимодействуют с гидроксильными ионами воды, образуя слабое основание. Суммарное уравнение гидролиза получается при сложении двух процессов:H2O  H+ + OH-B+ + OH- BOH B+ + H2O BOH + H+.
H+ + OH-B+ + OH- BOH B+ + H2O BOH + H+.
Уравнению гидролиза (14) отвечает выражение константы равновесия:
 , (15)
, (15)
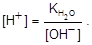 по которому вычисляют концентрацию водородных ионов.Допустим, что начальная концентрация этой соли равна СBA. Следовательно, соль полностью диссоциирует, согласно уравнению (13) [BA] = [B+] = [A-]. Из уравнения (14) следует, что [BOH] = [H+]. Концентрация негидролизованной части соли [B+] = СВА – [H+]. Так как [H+] <<CBA, принимают [B+] = CBA.Концентрацию ионов водорода находят из величины ионного произведения воды KH2O:
по которому вычисляют концентрацию водородных ионов.Допустим, что начальная концентрация этой соли равна СBA. Следовательно, соль полностью диссоциирует, согласно уравнению (13) [BA] = [B+] = [A-]. Из уравнения (14) следует, что [BOH] = [H+]. Концентрация негидролизованной части соли [B+] = СВА – [H+]. Так как [H+] <<CBA, принимают [B+] = CBA.Концентрацию ионов водорода находят из величины ионного произведения воды KH2O:
Концентрацию воды можно считать практически постоянной. Произведение K[H2O] называют константой гидролиза Kгидр.
Подставляя полученные выражения равновесных концентраций в уравнение (15), получим:
K[H2O] = Kгидр. =  или
или
K[H2O] = Kгидр.  ,
,
откуда Kгидр. 
тогда 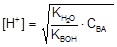 . (16)
. (16)
рН= -lg[Н+]
3) Ионный обмен — это обратимая химическая реакция, при которой происходит обмен ионами между твердым веществом (ионитом) и раствором электролита. Ионный обмен может происходить как в гомогенной среде (истинный раствор нескольких электролитов), так и в гетерогенной, в которой один из электролитов является твёрдым (при контакте раствора электролита с осадком, ионитом и др.).
Ионообменная хроматография является более частным вариантом ионной хроматографии. Этот вариант хроматографии позволяет разделять ионы и полярные молекулы на основании зарядов разделяемых молекул. Данный вид хроматографии позволяет разделить практически любые заряженные молекулы, в том числе: крупные — белки, малые—молекулы нуклеотидов и аминокислот. Часто ионообменную хроматографию используют как первый этап очистки белков. Для проведения ионообменной хроматографии применяют синтетические полимеры, чаще всего на основе полистирола и полифенолов. Группировки, обуславливающие ионообменные процессы, чаще всего вводят в качестве заместителей в бензольное кольцо мономеров. Выпускаемые промышленностью ионообменные смолы имеют вид небольших полимерных шариков, которые перед использованием нужно замачивать в воде или другом элюенте[1]. Набухание ионообменной смолы сопровождается увеличением доступности функциональных групп полимера за счет раздвигания макромолекул молекулами элюента, как при пластификации полимера. Степень набухания ионообменной смолы регулируют как длиной макромолекул, так и степенью поперечной сшивки полимерной матрицы.
В ионообменной хроматографии разделение компонентов смеси достигается за счет обратимого взаимодействия ионизирующихся веществ с ионными группами сорбента. В качестве подвижной фазы используют водные растворы солей кислот, оснований и растворители типа жидкого аммиака, т.е. системы растворителей, имеющих высокое значение диэлектрической проницаемости и большую тенденцию ионизировать соединения. Обычно работают с буферными растворами, дозволяющими регулировать значение рН.
Хроматофокусирование (ХФ) — метод ионообменной хроматографии, основанный на элюировании с внутренним (создаваемым внутри колонки) градиентом рН. В настоящее время используют две техники формирования градиентов рН – «классическое» хроматофокусирование и индуцирование. ХФ обычно применяют для разделения биологических объектов – белков, пептидов, аминокислот, реже азокрасителей и других цвиттер-ионных соединений.
Уравнение Никольского 
const – эмпирическая величина, определяемая для каждого ИСЭ, отрезок, на основании потенциалов, отсекаемый функцией;
+ в случае определения катионов, - в случае определения анионов;
zx – заряд определяемо иона, zy – заряд мешающего иона;
ax – активность определяемого иона, ау – активность мешающего иона;
Кх/у – коэффициент селективности определяемый ион - мешающий ион. Коэффициент селективности показывает, при каком избытке мешающего иона мы можем получить 100% погрешность в определении анализируемого иона.
Билет.
1) гидролиз солей – это процесс обменного разложения воды и растворенной в ней соли – электролита, приводящий к образованию малодиссоциирующего вещества. Характеризовать гидролиз количественно позволяют такие величины, как Степень гидролиза и константа гидролиза. Соли, образованные слабым основанием и сильной кислотой Такое соединение, при ионизации, образует катионы, способные к поляризации гидратной оболочки и анионы, которые их поляризуют слабо. Тогда гидроли з проходит по катиону, при этом среда носит кислый характер, т.е. рН ˂ 7: NH4Cl ↔ NH4+ + Cl—
NH4+ + HOH ↔ NH4OH + H+
Cl—+ HOH ↔ реакция практически не идет
NH4Cl+ HOH ↔ NH4OH + HCl
Для солей, образованных слабым основанием и сильной кислотой, константа гидролиза и константа диссоциации основания связаны соотношением: Kг = KH2O/Kосн
Понятно, что чем меньше сила основания, тем в большей степени протекает гидролиз.
Если соль образованна слабым основанием многовалентного металла и сильной кислотой, то ее гидролиз будет протекать ступенчато: FeCl2 ↔ Fe2+ + 2Cl—
| I ступень | Fe2++ HOH ↔ (FeOH)+ + H+FeCl2 + HOH ↔ (FeOH)Cl + HCl |
| 2ступень | (FeOH)+ + HOH ↔ Fe(OH)2 + H+(FeOH)Cl + HOH↔ Fe(OH)2 + HCl |
Константа гидролиза по первой ступени связана с константой диссоциации основания по второй ступени, а константа гидролиза по второй ступени — с константой диссоциации основания по первой ступени: Kг1 = KH2O/Kосн2 Kг2 = KH2O/Kосн1
Поскольку первая константа диссоциации кислоты всегда больше второй, то первая константа гидролиза всегда больше, чем константа вторая гидролиза, так как первая константа диссоциации основания всегда больше второй Kг1> Kг2
Гидролиз всегда будет протекать в большей степени, чем по второй. Этому также способствуют ионы, которые образуются при гидролизе по первой ступени, они приводят подавлению гидролиза по второй ступени, смещая равновесие влево.
Сравнивая величины Kг и Kосн можно качественно определить pH среды. Так, если Kг намного больше Kосн, то среда сильнокислая, при Kг намного меньшей Kосн — среда слабокислая, а если Kг и Kосн сопоставимы, то — среднекислая.
2) В меркуриметрии используют образование галогенидных и псевдогалогенидных комплексов Hg(II). В условиях проведения титрования протекает реакция: 2Cl- + Hg2+ = HgCl2.
Аналогичные реакции протекают с бромид-, йодид-, роданид- и цианид-ионами; можно определять также соли Hg(II). HgCl2- малодиссоциированная соль. Титрант: вторичный стандартный раствор Hg(NO3)2. Стандартизация: за первичным стандартным раствором натрий хлорида NaCl: Hg(NO3)2 + 2NaCl = HgCl2 + 2NaNO3. В качестве индикатора (натрий нитропруссид) Na2[Fe(CN)5NO], который образует с Hg2+-ионами малорастворимую белую соль:
Na2[Fe(CN)5NO] + Hg(NO3)2 = 2NaNO3 + Hg[Fe(CN)5NO]¯белыйосадок.
Значительно удобнее вести титрование с раствором дифенилкарбазона в качестве индикатора, который образует с ионами Hg2+ осадок интенсивно синего цвета.
Меркуриметрическое определение йодидов базируется на такой реакции: Hg2++ 4I- = [HgI4]2-.
 В точке эквивалентности ионы Hg2+ реагируют с ионами тетрайодомеркурата (ІІ) [HgI4]2-, образуя осадок йодида меркурия (ІІ) красного цвета: HgI42- + Hg2+ = HgI2¯. При определении роданидов (тиоцианатов SCN-), как индикатор используют раствор соли Fe(III), которые образуют с роданид-ионами комплексное соединение красного цвета: Fe3+ +3SCN- = Fe(SCN)3. В процессе титрования ионы Hg2+ связывают SCN --ионы: Hg2+ + 2SCN- = Hg(SCN)2, После полного оттитровывания свободных тиоцианат-ионов с титрантом начинает реагировать тиоцианатные комплексы ферума (ІІІ), в результате чего красная окраска Fe(SCN)3 исчезает, что является признаком конца титрования: 2Fe(SCN)3 + 3Hg(NO3)2 = 3Hg(SCN)2 + 2Fe(NO3)3. Аналогично определяют хорошо диссоциированные соли меркурия (ІІ), титруя их раствором калий тиоцианата в присутствии ионов Fe(III). В точке эквивалентности возникает красная окраска ферум (ІІІ) тиоцианата:Hg2+ + 2SCN- = Hg(SCN)2; Fe3+ + 3SCN- = Fe(SCN)3. Наибольшее практическое значение имеет определение хлоридов. Хорошие результаты при этом получают даже при таких маленьких концентрациях хлоридов, которые встречаются в питьевой воде. Бромид-, цианид-, и роданид-ионы можно определять аналогично, а титрование йодид-ионов следует проводить в присутствии этанола для снижения растворимости HgІ2, который образуется. Главный недостаток меркуриметрии - высокая токсичность соединений меркурия.
В точке эквивалентности ионы Hg2+ реагируют с ионами тетрайодомеркурата (ІІ) [HgI4]2-, образуя осадок йодида меркурия (ІІ) красного цвета: HgI42- + Hg2+ = HgI2¯. При определении роданидов (тиоцианатов SCN-), как индикатор используют раствор соли Fe(III), которые образуют с роданид-ионами комплексное соединение красного цвета: Fe3+ +3SCN- = Fe(SCN)3. В процессе титрования ионы Hg2+ связывают SCN --ионы: Hg2+ + 2SCN- = Hg(SCN)2, После полного оттитровывания свободных тиоцианат-ионов с титрантом начинает реагировать тиоцианатные комплексы ферума (ІІІ), в результате чего красная окраска Fe(SCN)3 исчезает, что является признаком конца титрования: 2Fe(SCN)3 + 3Hg(NO3)2 = 3Hg(SCN)2 + 2Fe(NO3)3. Аналогично определяют хорошо диссоциированные соли меркурия (ІІ), титруя их раствором калий тиоцианата в присутствии ионов Fe(III). В точке эквивалентности возникает красная окраска ферум (ІІІ) тиоцианата:Hg2+ + 2SCN- = Hg(SCN)2; Fe3+ + 3SCN- = Fe(SCN)3. Наибольшее практическое значение имеет определение хлоридов. Хорошие результаты при этом получают даже при таких маленьких концентрациях хлоридов, которые встречаются в питьевой воде. Бромид-, цианид-, и роданид-ионы можно определять аналогично, а титрование йодид-ионов следует проводить в присутствии этанола для снижения растворимости HgІ2, который образуется. Главный недостаток меркуриметрии - высокая токсичность соединений меркурия.
3) Нефелометрия — метод исследования и анализа вещества по интенсивности светового потока, рассеиваемого взвешенными частицами данного вещества.
Интенсивность рассеянного светового потока зависит от множества факторов, в частности от концентрации частиц в анализируемой пробе. Большое значение при нефелометрии имеет объём частиц, рассеивающих свет. Важное требование к реакциям, применяемым при нефелометрии, заключается в том, что продукт реакции должен быть практически нерастворим и представлять собой суспензию (взвесь). Для удержания твёрдых частиц во взвешенном состоянии применяются различные стабилизаторы (например, желатин), предотвращающие коагуляцию частиц.
Для измерения интенсивности рассеянного света используются специальные приборы — нефелометры. Их действие основано на уравнивании двух световых потоков: одного от рассеивающей взвеси, другого от матового или молочного стеклянного рассеивателя прибора. Один из вариантов нефелометрии — нефелометрическое титрование, в котором раствор анализируемого вещества титруют раствором осадителя. В процессе титрования интенсивность рассеянного света увеличивается пропорционально количеству образующихся частиц. В точке эквивалентности рост помутнения прекращается. По излому кривой титрования находят объём затраченного на реакцию осадителя. Погрешность при этом составляет от 5 до 10 %.
Нефелометрия используется преимущественно для определения хлоридов (в виде взвеси AgCl), сульфатов (в виде взвеси BaSO4) при анализе различных материалов, например руд, минералов. Нефелометрию также применяют для определения размеров и формы диспергированных частиц, молекулярной массыполимеров, изучения коагуляции.
фотонефелометрия основана на явлении рассеяния света дисперсными системами — суспензиями или золями, получаемыми в результате аналитических реакций. При прохождении света через дисперсную гетерогенную систему, какой является взвесь малорастворимого вещества в момент образования, происходит ослабление светового потока в результате рассеивания и поглощения его частицами дисперсной фазы.
Закон Рэлея — Джинса — закон излучения для равновесной плотности излученияu (ω, T) {\displaystyle u(\omega,T)} и для испускательной способностиf (ω, T) {\displaystyle f(\omega,T)} абсолютно чёрного тела, который получили Рэлей и Джинс в рамках классической статистики (теорема о равнораспределении энергии по степеням свободы и представление об электромагнитном поле как о бесконечномерной динамической системе) ФОТОНЕФЕЛОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ.как только будет достигнута точка эквивалентности, индикатор обесцветится, лучи начнут проходить через раствор без поглощения и сила фототока в фотоэлементе резко увеличится. Точку эквивалентности устанавливают по показаниям специальных приборов либо с помощью индикаторов. Момент, при котором происходит изменение цвета индикатора, называют точкой конца титрования. Они используются для приготовления стандартных (титрованных) растворов, как индикаторы, маскировочные средства, осадители и соосадители и т.
БИЛЕТ.
1)Гидролитическое равновесие в водных растворах солей следует закону действующих масс. Изменяя концентрацию одного из продуктов реакции, можно смещать равновесие в одну или иную сторону. Гидролитическое равновесие в разбавленных растворах соли характеризуется константой гидролиза. Константа гидролиза — константа равновесия гидролитической реакции. Так константа гидролиза соли равна отношению произведения равновесных концентраций продуктов реакции гидролиза к равновесной концентрации соли с учетом стехиометрических коэффициентов. Гидролитическое равновесие, наступающее в результате реакции, зависит от концентрации и температуры. С повышением температуры и с увеличением разведения степень гидролиза возрастает. Этим пользуются при получении золя гидрата окиси железа.В качестве примера ниже приводится вывод уравнения константы гидролиза соли, образованной слабой кислотой и сильным основанием:
N a N O 2 + H 2 O ⇌ H N O 2 + N a O H {\displaystyle NaNO_{2}+H_{2}O\rightleftharpoons HNO_{2}+NaOH} 2) Метод Фольгарда – это метод обратного титрования применяют для количественного определения хлоридов, бромидов, йодидов в азотнокислой среде. Рабочими растворами являются р-р AgNO3 0,1М и раствор NH4CNS 0,1М. Индикатор – FeNH4(SО4)2 – железоаммониевые квасцы. К раствору точной навески галогенида прибавляют полуторный или двойной избыток титрованного раствора нитрата серебра, разведенной азотной кислоты, 10-20 капель индикатора (железоаммониевые квасцы) и титруют избыток нитрата серебра раствором роданида аммония до буровато-оранжевого окрашивания раствора над осадком, устойчивого при непродолжительном вращательном движении. В данном методе часть AgNO3 реагирует с ионами галогена, образуя осадок галогенидов серебра. А остальная часть оттитровывается NH4CNS с образованием роданида серебра AgCNS. После связывания ионов серебра лишняя капля NH4CNS будет реагировать с железоаммониевыми квасцами с образованием буровато-оранжевого окрашивания раствора Fe(CNS)3, что указывает на достижение точки эквивалентности.
KJ + AgNO3 ® ¯AgJ +KNO3
AgNO3 + NH4CNS ® ¯AgCNS + NH4NO3
3NH4CNS + FeNH4(SO4)2 ® Fe(CNS)3 +2(NH4)SO4
Количество AgNO3, которое пошло на взаимодействие с галогенидом определяют как разность между взятым количеством AgNO3 и оставшимся в избытке. Преимущества: метод применим для определения хлоридов, бромидов, иодидов, роданидов и ионов серебра. Метод применим для титрования кислых растворов, так как осадок нерастворим в кислотах. Эта особенность метода делает его очень удобным при анализе серебряных сплавов, которые растворяют в кислотах, и количественном определении галогенидов в сильнокислых средах, так как галогениды в указанных средах нельзя титровать по методу Мора или в присутствии адсорбционных индикаторов недостатков метода является использование дорогостоящих солей серебра.
3) Физико-химические методы анализа основаны на том, что анализируемый объект подвергают химическим превращениям, при этом измеряют физико-химические свойства вещества (аналитический сигнал), которые регистрируются приборами. Аналитический сигнал Хiзависит от природы и концентрации определяемого вещества.
Физические методы анализа основаны на измерении физических характеристик, которые зависят от индивидуальных свойств вещества (показатель преломления, теплопроводность и т. д.). В этих методах не применяются химические или электрохимические реакции.При выполнении анализов физико-химическими методами точку эквивалентности (конец реакции) определяют не визуально, а при помощи приборов, которые фиксируют изменение физических свойств исследуемого вещества в точке эквивалентности. Для этой цели обычно применяют приборы с относительно сложными оптическими или электрическими схемами, поэтому эти методы получили название методов инструментального анализа. Среди физико-химических методов наибольшее практическое применение имеют: 1. спектральные и другие оптические методы (рефрактометрия, поляриметрия);2. электрохимические методы анализа;3. хроматографические методы анализа. На измерении эффектов поляризации молекул вещества основаны рефрактометрия, поляриметрия. Анализируемые вещества могут поглощать электромагнитное излучение и на основе использования этого явления выделяют группу абсорбционных оптических методов. Поглощение света атомами анализируемых веществ используется в атомно-абсорбционном анализе. Способность поглощать свет молекулами и ионами в ультрафиолетовой, видимой и инфракрасной областях спектра позволила создать молекулярно-абсорбционный анализ (колориметрию, фотоколориметрию, спектрофотометрию). Поглощение и рассеяние света взвешенными частицами в растворе (суспензии) привело к появлению методов турбидиметрии и нефелометрии. Методы, основанные на измерении интенсивности излучения, возникающего в результате выделения энергии возбужденными молекулами и атомами анализируемого вещества, называются эмиссионными методами. К молекулярно-эмиссионным методам относят люминесценцию (флуоресценцию), к атомно-эмиссионным - эмиссионный спектральный анализ и пламенную фотометрию. Электрохимические методы анализа основаны на измерении электрической проводимости (кондуктометрия); разности потенциалов (потенциометрия); количества электричества, прошедшего через раствор (кулонометрия); зависимости величины тока от приложенного потенциала (вольт-амперометрия). В группу хроматографических методов анализа входят методы газовой и газожидкостной хроматографии, распределительной, тонкослойной, адсорбционной, ионообменной и других видов хроматографии. Плюсы: Низкий предел обнаружения (10-3-10-12 г) и малая предельная концентрация (до ~ 10-12 г/мл) определяемого вещества. Высокая чувствительность; Высокая селективность (избирательность) методов. Минусы:Воспроизводимость результатов часто ниже, чем при использовании классических химических методов количественного анализа (гравиметрия, титриметрия). Большая погрешность определений, составляющая 5 – 20%, в то время как в классическом химическом анализе (гравиметрия, титриметрия) погрешность определений не превышает 0,1 – 0,5%. Сложность используемой аппаратуры, ее высокая стоимость.
Билет № 26
1. Соль сильной кислоты и сильного основания гидролизу не подвергается, рН = 7
Соль слабой кислоты и сильного основания рН > 7
Пример: ацетат натрия CH3COONa (см.ниже)
Соль слабой кислоты и слабого основания – рН зависит от соотношения констант диссоциации кислоты и соли. Иногда происходит необратимый гидролиз.
CH3COONa + H2O Û CH3COOH + NaOH
CH3COO– + H2O Û CH3COOH + OH–
«константа гидролиза» Кг = ([AН] [OH–]) /[A–]
В нем мы можем [A–] приравнять к концентрации исходной соли (2 М), а [AН] и [OH–] считать равными. Тогда: Кг = ([OH–]2) /[2 М] Но табличных данных по Кг нет. Тогда выражение Кг = ([AН] [OH–]) /[A–] умножаем и делим на [H+]: Кг = ([AН] [OH–][H+]) /([A–][H+]) При этом оно сводится к двум известным константам: Кг = Kw / Ккисл = 10-14/1,8*10-5В результате [OH–] = Ö (Кг 2 М) = Ö [(10-14/1,8*10-5)(2 М)]»Ö 10-9 = 10-4,5Тогда рОН = 4,5; рН = 14 – рОН = 9,5
2. Аргентометрия основана на применении в качестве осадителя стандартного раствора серебра нитрата:
Аg+ + Наl- ↔ АgНаl↓ Стандартный раствор 0,1 моль/дм3серебра нитрата может быть приготовлен:
-как первичный стандартный раствор; - вторичный стандартный раствор.
Для приготовления первичного стандартного 0,1 моль/дм3 раствора AgNО3 рассчитанную навеску химически чистой соли AgNО3 взвешивают на аналитических весах, переносят в мерную колбу, растворяют в дистиллированной воде, доводят объем раствора до метки, тщательно перемешивают и переносят в склянку из темного стекла. При приготовлении вторичног о стандартного раствора АgNО3рассчитанную навеску соли взвешивают на технических весах, переносят через воронку в склянку из темного стекла, добавляют цилиндром необходимый объем дистиллированной воды и тщательно перемешивают. Полученный вторичный стандартный раствор AgNО3 стандартизуют по химически чистым стандартным веществам KCl или NaCl или же по их растворам. Концентрация стандартных растворов серебра нитрата изменяется при длительном хранении. Причиной нестойкости растворов серебра нитрата является их светочувствительность, потому эти растворы следует хранить в склянках из темного стекла либо в посуде, обернутой черной бумагой или покрытой черным лаком, и в защищенном от света месте. Их концентрацию необходимо периодически проверять. Способы определения конечной точки титрования Безындикаторные способы лорид - ионы определяют по так называемому способу равного помутнения (метод Гей-Люссака). При этом анализируемый раствор титруют стандартным раствором серебра нитрата, конец титрования определяют путем отбора двух проб титруемого раствора в две пробирки вблизи конечной точки титрования: в одну из них прибавляют каплю стандартного раствора серебра нитрата, в другую – каплю стандартного раствора натрия хлорида такой же концентрации. В недотитрованном растворе появляется помутнение в пробирке с серебра нитратом, в перетитрованном - в пробирке с натрия хлоридом. В конечной точке титрования раствор в обеих пробирках имеет одинаковое помутнение. Бромид- и йодид- ионы определяют безындикаторным способом просветления. Суть его состоит в том, что при добавлении к анализируемому раствору из бюретки небольшими порциями стандартного раствора серебра нитрата в начале образуется коллоидный раствор серебра бромида, а в момент эквивалентности происходят коагуляция коллоидных частиц и осаждение их в виде творожистых хлопьев, раствор при этом осветляется. Этот метод достаточно точен, но в настоящее время применяется редко. Из современных безындикаторных методов в аргентометрии чаще всего применяется потенциометрическое определение точки эквивалентности с использованием серебряных или галогенид-селективных электродов. Индикаторные способы: метод Мора, основанный на реакции между ионами серебра и галогенид - ионами в присутствии индикатора - раствора калия хромата; - метод Фольгарда (тиоцианатометрия), основанный на реакции между ионами серебра и тиоцианат-ионами в присутствии ионов железа (III) в качестве индикатора; - метод Фаянса - Ходакова основан на применении адсорбционных индикаторов.
3.РЕФРАКТОМЕТРИЯ - метод исследования в-в, основанный на определении показателя преломления (коэф. рефракции) и нек-рых его ф-ций. Показатель преломления n -отношение скоростей света в граничащих средах. Для жидкостей и твердых тел n определяют, как правило, относительно воздуха, для газов -относительно вакуума. Значения n зависят от длины волны l света и т-ры, напр.  -показатель преломления при 20 °С для D-линии спектра натрия. Часто используют также линии С и F спектра водорода В случае газов необходимо учитывать зависимость n отдавления (указывать его или приводить данные к нормальному давлению). Анизотропные тела-одно- и двухосные кристаллы-характеризуются соотв. двумя экстремальными или тремя значениями n.
-показатель преломления при 20 °С для D-линии спектра натрия. Часто используют также линии С и F спектра водорода В случае газов необходимо учитывать зависимость n отдавления (указывать его или приводить данные к нормальному давлению). Анизотропные тела-одно- и двухосные кристаллы-характеризуются соотв. двумя экстремальными или тремя значениями n.
Обычно n жидких и твердых тел определяют с точностью до 0,0001 на рефрактометрах, в к-рых измеряют предельные углы полного внутр. отражения; при этом нет необходимости придавать образцу строго определенную геом. форму. Наиб.распространены рефрактометры с призменными блоками и компенсаторами дисперсии Аббе, позволяющие определять nD в "белом" свете по шкале или цифровомуиндикатору. Интерферометры используют также для точного (до 10-7) определения разностей n р-ров. Для этой же цели служат дифференц. рефрактометры, основанные на отклонении лучей системой двух-трех полых призм. Приидентификации минералов n мелких крупинок (порошков) определяют иммерсионным методом, погружая крупинки в капли иммерсионных жидкостей с известными n и наблюдая в микроскоп (иногда при нагр. или изменении длины волны света) момент совпадения п. Обратный вариант иммерсионного метода-идентификация расплавов орг. в-в с помощью микроскопа и набора стеклянных порошков с известными n (метод Кофлера)-получил распространение при анализе лек. препаратов. Из ф-ций и, используемых в химии, наиб. значение имеют: ф-ция Лоренца-Лоренца, производная n поконцентрации растворенных в-в (инкремент п)и дисперсионные ф-лы, включающие разности показателей преломления для двух длин волн. Инкременты n используют в жидкостной хроматографии и при определении мол.массы полимеров методом рассеяния света. Для рефрактометрич. анализа р-ров в широких диапазонах концентраций пользуются таблицами или эмпирич. ф-лами, важнейшие из к-рых (для р-ров сахарозы, этанола и др.) утверждаются международными соглашениями и лежат в основе построения шкал специализир. рефрактометров для анализа пром. и с.-х. продукции. Разработаны способы анализа трехкомпонентных р-ров, основанные на одновременном определении их n и плотности или вязкости, либо на проведении хим. превращений с измерением n исходных и конечных р-ров; эти способы применяют при контроле нефтепродуктов, фармацевтич. препаратов и др. Идентификация орг. соединений, минералов, лек. в-в осуществляется по таблицам п, приводимым в справочных изданиях.
Билет № 27
1.


2. Метод Фаянса – это метод прямого титрования галогенидов раствором AgNO3 0,1М в слабо кислой среде с применением адсорбционных индикаторов, которые показывают изменение цвета не в растворах, а на поверхности выпавшего осадка. индикаторов 1. Бромфеноловый синий, бромкрезеловый синий – в уксуснокислой среде;2. Эозинат натрия – в уксуснокислой среде; 3. Флуоресцеин – в нейтральной и слабо щелочной среде. Хлориды и бромиды можно титровать с бромфеноловым синим. Точную навеску хлорида или бромида растворяют в воде, прибавляют 2-3 капли индикатора бромфенолового синего, по каплям разведенную уксусную кислоту до зелено-желтого окрашивания и раствор AgNO3 0,1М до синего окрашивания. Можно с этим индикатором оттитровать и йодиды, только вместо синего окрашивания в точке эквивалентности будет зеленое окрашивание. В качестве индикатора для определения йодидов применяют эозинат натрия. К точной навеске препарата прибавляют разведенную уксусную кислоту, 3-5 капель раствора эозината натрия и титруют раствором AgNO3 0,1М до розового окрашивания осадка. Определению йодидов методом Фаянса не мешают хлориды, но мешают бромиды. Примечание. Методом определяют соли алкалоидов, соли азотосодержащих оснований (димедрол, новокаин, папаверин, дикаин, пилокарпил, эфедрин). применяетс я для определения концентрации йодидов, но он может использоваться также для хлоридов и бромидов. В отличие от метода Мора, титрование выполняется не только в нейтральной среде, но и в среде уксусной кислоты с водным раствором эозината натрия в качестве индикатора. В точке эквивалентности наблюдается появление ярко-розового окрашинивания осадка. Хлориды и бромиды титруютв среде уксусной кислоты, индикатором служит раствор бром-фенолового синего. В точке эквивалентности зеленовато-желтое окрашивание переходит в сине- фиолетовое.
3.Колориме́трия») — физический метод химического анализа, основанный на определении концентрации вещества по интенсивности окраски растворов (более точно — по поглощению света растворами). Колориметрия — это метод количественного определения содержания веществ в растворах, либо визуально, либо с помощью приборов, таких как колориметры. Колориметрия может быть использована для количественного определения всех тех веществ, которые дают окрашенные растворы, или могут дать окрашенное растворимое соединение с помощью химической реакции. Колориметрические методы основываются на сравнении интенсивности окраски исследуемого раствора, изучаемого в пропущенном свете, с окраской эталонного раствора, содержащего строго определенное количество этого же окрашенного вещества, или же с дистиллированной водой. Более совершенные приборы — спектрофотометры — отличаются возможностью исследования оптической плотности в широком диапазоне длин волн видимого спектра, а также в ИК и УФ-диапазонах, с меньшей дискретностью длины волны (с использованием монохроматора). Фотоколориметры и спектрофотометры измеряют величину пропускания света при определенной длине волны света. Контроль (обычно дистиллированная вода или исходный материал без добавления реагентов) используется для калибровки устройства применяется в аналитической химии, в том числе для гидрохимического анализа, в частности — для количественного анализа содержания биогенных веществ в природных вода, для измерения pH, в медицине, а также в промышленности при контроле качества продукции.
Билет № 28
1.Анал р-я - Хим р-я, сопр-ся анал сигналом (признаком), по которому можно судить о наличии определяемого вещества.

