2018-01-21
2018-01-21 928
928Молекулярность – количество частиц, принимающих участие в элементарном акте хим превращения.Реакции могут быть моно-, би-, тримолекулярными (не больше). Мономолекулярные-реакции разложения и внутримолек пергруппировки (J2)=2(J). Бимолекулярные-2NO2=N2O4. Тримолекулярные(рекдие) 2NO+O2=2NO2. При комнатной температуре ср скорость движ частиц такова, чтоподойдя близко друг к другу, они испытывают отталкивание эл облаков и расходятся в разные стороны-упругое столкновение. Только те частицы, у которых Екин достаточно велика, чтобы преодолеть отталкивание, провзаимоде ствуют. Некоторые реакции, которые должны идти по термодин сообр, при Тк «заморожены». Энергия активации-избыточная энергия, кот должны обладать молекулы во время столкновения, чтобы произошло взаимодействие. Число столкновений пропроционально Т1/2, т.е. при ΔТ=1000 скорость реакции должна была бы возрасти в n=1.2 раза. Но по Вант-Гоффу она возр минимум в 210 раз. Распределение Максвелла – Больцмана – распределние частиц по энергии или по скоростям Nакт=Nобще-E/RT. е=2.718. Отсюда Аррениус преложил формулу для зависимости конст скорости реакции от температуры k=Ae-Ea/RT. Еа-энергия активации, кот нужна для начала процесса, но не влияет на ΔН. А – стерический фактор, кот опред вероятность активного столкновения. Если сталкиваются сложные молекулы, они должны подойти друг к жругу опред концами. Напр: при окислении NO кислород должен подойти к азоту. Пример: столкновение Н2 и J2. (H2) + (I2) = 2(HI) DН=+58.2 кДж Еа= 168 кДж сумма энергий связи в молекулах водорода и иода 571, следоват связи не рвутся. Еа зависит от механизма реакции: 1) простые (H2) + (I2) = 2(HI) Еа= 150-450 кДж/моль 2)реакции ионов с молекулами Еа= 0-80 кДж/моль. Пример: облучение светом молекулы воды ионизирует ее. 3) радикальные реакции – во взаимод вступают радикалы – молекулы с несп эл. ·ОН, · NH2. Еа=0-40 кДж/моль. Частный случай – цепные реакции.
Катализ.
Катализатор-вещ-во, ускоряющеереакцию, но не участвующее в ней. Катализатор участвует в реакции, проводя ее по другому пути, которому отвечает меньшая эенргия акт или ориентирует молекулы соотв образом, присоединяя их к себе. Выглядит катализ: А+К=А*, А*+В=С+D+К. Катализ может быть гетерогенным и гомогенным. Пример гомогенного: окисление SO2+1/2O2=SO3 в присутствии NO. NO легко окисляется до NO2, а NO2 окисляет SO2. Гетерогенный катализ: 2Н2О2=2Н2О+1/2О2 (катализатор MnO2, в живых орг - каталаза). Специфичность действия катализатора – продукты реакции могут быть разными в зависимости от того, какой катализатор исп. Как реально происходит катализ: А+К→АК – неустойчив, АК+В→АКВ – переходный комплекс. Пример: омыление эфира аминоуксусной к-ты – влияние катализатора на эл плотность.Ионы меди образ комплекс. Гетерогенный катализ удобен тем, что не надо отделять продукты реакции от катализатора. Катализ происходит на поверхности за счет хемосорбции. Пример: синтез аммиака на железе. 3 стадии 1)диффузия 2)адсорбция 3)десорбция. 1) Н2+2[ ]↔2[H] 2) N2+2[ ]↔2[N] лимитирующая стадия 3) [N]+[H]→[NH] + [ ] 4) [NH]+[H]→[NH2]+[ ] 5)[NH2]+[H]→[NH3]+[ ] 6) [NH3]↔NH3 + [ ]. Теплота хемосорбции должна быть достаточно велика, но не очень, иначе продукты не будут десорбироваться. Большую роль в гетерогенном катализе играют процессы переноса.
Равновесие.
Обратимые-реакции, способные идти в двух противоположных направлениях. Рассм обр реакцию А+В=D+F. В начальный момент времени, когда СА и СВ максимальны, скорость прямой реакции тоже максимальна. С течением времени скорость прямой реакции падет Vпр=кпр[A][B]. Реакция приводит к образованию D и F,молекулы кот могут вновь реагировать, давая А и В. Чем выше конц D и F, тем вероятнее обратный процесс, тем выше скорость обратной реакции. Vоб=kоб[D][F]. Изменение скоростей прямой и обратной реакций можно предс графиком. по мере прохождения реакции наст такой момент, когда скорости пр и обр реакций делаются равными, кривые Vпр и Vоб сливаются в одну прямую, параллельную оси t. Такое сост-равновесие. При равновесии конц всех участников реакции ост пост и не меняются со временем, хотя одновр идут и пр и обр реакции. Т.е. равновесие явл динамическим, это доказал Боденштерн. В равновесную смесь Н2+J2↔2HJ он добавлял радиоактивный иод, который вскоре обнаруживался в пробах HJ. При равновесии Vпр= Vобр или kпр[A][B]=kоб[D][F]. Откуда [D][F]/[A][B]=K=kпр/kобр – константа хим равновесия. Если реакции характ наличием отличных от 1 коэфф aA + bB «dD + fF. Каждую сложную реакцию можно представить в виде ряда последов простых реакций, для них можно записать соотв кинетич уравнения для прямой и обр реакции и выражения для конст равнов. При суммировании последоват реакций их конс равнов пермнож и в суммарное выраж для К равно конц войдут в степенях равных стехиометр коэфф. К=[D]d[F]f/[A]a[B]b. Для одной и той же температуры отношение произведений равновесных конц (в степенях их стехиом коэфф) вещ-в в правой и левой частях ур-ния хим реакции представл пост величину – закон действ масс. 1) все это верно для равнов конц 2)записывая конст, в числ стоят равнов конц кон прод, а в знам – нач вещ-в (так договорились). К показывает глубину протекания процесса. Если К>>1, процесс сильно сдвинут в сторону получ прод реакции. Если К<<1, то процесс сдвинут влево и практич не идет. К=1 – равновесие установилось на «полдороге». Различают истинное и мнимое равновесие, которое назыв заторможенным равнов или метастаб сост (в колбе водород, кислород, вода – ничего годами не менятеся, чтобы понять, что это не равнов надо поднести спичку). Признаки истинного равн: 1)при сохр внешних усл сост системы не меняется во времени 2)при изм усл (введениие доп количеств реаг вещ-в, изм давл или темп) система приходит к новому сост равнов. 3)к сост равнов можно подойти с противоп сторон. Равновесие можно сдвигать, меняя условия. Принцип Ле-Шателье: если на систему, находящуюся в рановесии, оказывается воздействие, равновесие смещ в сторону той реакции, которая ослабл это возд. Пример: 2SO2+O2↔2SO3 увелич конц кислорода приведет к смещ равнов вправо. Повыш темп смещ равно в сторону эндотерм реакции, т.к. при этом поголщ избыт тепло и темп пониж. CaCO3↔CaO+CO2 ΔH= +178кДж – повыш темп смещ равнов в сторону разлож карбоната. При увели давл равнов смещ в сторону уменьш количетсва молей газа. Для равнов между газообр вещ-ми удобно пользов не мол конц, а парц давл. Константа равнов, выраж через молярные конц – Кс, а через парц давл Кр. Связь между Кс и Кр на примере синтеза аммиака N2+3H2↔2NH3 Кс=[NH3]2/[N2][H2]3. Конц выражены в числе молей на литр смеси, т.е С=n/V. По уравнению Менд-Клайп pV=nRT →n/V=p/RT. подставим в уравн Кс. Получаем Кс=Кр(RT)Δn, где Δn – разность числа молей исх вещ-в и кон прод. Конст равнов опред путем анализа равнов смеси.
 Энтропия.
Энтропия.
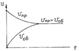
Осн вопрос- принципиальная возм самопроизв протекания процесса, его напр. Бертло и Томсен сформ принцип: любой хим процесс должен сопр выд тепла. Раньше думали, что все теплоты образов отриц, но выяснили, что бывают и полож, напр теплота образов оксидов азота. Самопроизв идут многие эндотерм реакции, напр растворение солей.следоват критерия Бертло и Томсона не достаточно. Пример: два сосуда с разными газами, откроем кран, газы смешаются, изм внутр энергии не происх, но процесс идет самопроизв, хотя на их разделение потребуется затрата работы. Все это потому что изм порядок. Самопроизв процесс, проходящий без изм энтальпии, соверш в направл, при котором беспорядок в сист возраст. Т.к. смешение газов более вероятно, чем их разделение, можно говорить, что движущей силой смешения газов явл тенденция перейти в более вероятн сост. Энергетич запасы смешив газов складываются, а вероятности сост перемножаются, для опред процесса нужно суммировать две движущ силы. S=RlnW – энтропия. W-вероятности. Энтропия – логарифмич выраж вероятности существ системы = мера беспорядка в системе. Изм в Дж/К*моль. Процесс, проходящий без изменения энергетич сост сист, совершается самопроизв только в напр, при кот энтропия сист возраст. Энтропия жидкости превыш энтропию тв тела, а энтропия газа-энтропию жидкости. При одинак усл в каждый момент врем получим разное распол молекул, т.е. множество микросост, кот нельзя наложить друг на друга так, чтобы они совпад.Макросост опред темп и давл, а микросост – число степеней свободы.Одноатомный газ-три степени свободы частиц – движ в трехмерном простр. Двухат газ- добавл врщат степ свободы и колеб атомов. Чем сложнее молекула газа,тем больше ее энтропия. При абсолютном 0 темп любое вещ-во должно предст собой идеальный кристалл. Вероятнось такого сост =1, а энтропия=0. Постулат Нернста-при абсолютном 0 энтропия соверш кристалла =0(3е начало теродинамики). 1можно гов об абсолютном знач энтропии 2)о завис энтропии от темп. При Т=0 S=0. При повы Т начин колеб атомов и S растет до Тпл. с повыш темп S плвно и незначит растет до Тисп, где резкий скачок ΔSисп и опять плвное увелич. Для энтроп хар з Гесса – изм энтропии не зависит от путипроцесса, а зав только от нач и кон сост. ΔS=∑Sкон-∑Sнач. Уменьш кол-ва газовых моей означ уменьш энтропии и наоб. Зависит от конц. S=S0-RlnC, S0-станд энтропия.