 2017-11-01
2017-11-01 2103
2103Термодинамические потенциалы идеальных газов
Как уже отмечалось, в идеальном газе отсутствуют силы межмолекулярного взаимодействия, а уравнением состояния является уравнение Менделеева – Клапейрона
pV = nRT, (5.1)
где R – универсальная газовая постоянная, представляющая собой работу расширения газа при повышении температуры на 1 К, n – число молей газа.
Так как внутренняя энергия идеального газа не зависит от объема и давления, а является лишь функцией температуры, то на основании уравнения (2.7) после его интегрирования получим для внутренней энергии при любой температуре T:
UТ = U о + сV (Т – Т o). (5.2)
Энтальпия идеального газа, как и внутренняя энергия, также зависит только от температуры. Так как, по определению, H = U + pV, а для 1 моля газа pV = RT и (ср – сV) = R, то
Н = U о + ср (Т – Т o). (5.3)
Изохорный и изобарный потенциалы идеального газа при постоянной температуре определяются интегрированием уравнений (4.28) и (4.34). Для одного моля идеального газа при T = const
dF = – pdV = –  dV, (5.4)
dV, (5.4)
откуда после интегрирования получаем
F = F (Т) – RT ln V. (5.5)
Для изобарного потенциала
dG = Vdp =  dp, (5.6)
dp, (5.6)
откуда
G = G (Т) + RT ln p. (5.7)
Величины F (T) и G (T) в уравнениях (5.5) и (5.7) представляют собой константы интегрирования, зависящие от температуры, но их значения неизвестны, а поэтому и величины F и G остаются неопределенными. Однако на практике обычно важно знать не абсолютные значения термодинамических потенциалов, а их изменения в том или ином процессе, и приведенные уравнения позволяют рассчитывать эти изменения. Например, при изотермическом расширении (сжатии) моля идеального газа изменение изобарного потенциала будет равно
D G = G 2 – G 1 = G (Т) + RT ln p 2 – G (Т) – RT ln p 1 = RT ln(р 2/ р 1). (5.8)
Постоянные G (T) сокращаются, так как при заданной температуре они одинаковы при любых давлениях.
Реальные газы. Летучесть
Уравнения для термодинамических потенциалов реальных газов также можно, в принципе, получить на основании уравнений состояния. Однако в действительности мы не имеем уравнения состояния реального газа, которое описывало бы связь между параметрами состояния при любых условиях. Единственным теоретически обоснованным уравнением для реальных газов является уравнение совторым вириальным коэффициентом
pV = RT (1 + В / V), (5.9)
где
В = В о + a / Тn – b/Тm. (5.10)
 Рис. 5.1. Изотермы реальных газов (Т 1< Т 2 < Ткр < Т 3) Рис. 5.1. Изотермы реальных газов (Т 1< Т 2 < Ткр < Т 3) |
Это уравнение выведено методами статистической механики и имеет большое теоретическое значение, но не представляет существенного практического интереса вследствие сложности определения B и правильности уравнения только при малых отклонениях от законов идеального газа.
Широко известно уравнение Ван-дер-Ваальса:
(р + а / V 2)(V –b) = RT, (5.11)
которое характеризует не только изменения состояния газа, но и жидкости. Постоянные a и b для каждого газа вычисляются из его критических параметров. Однако количественно это уравнение не всегда отвечает экспериментальным данным, а опытные значения a и b в обычных условиях примерно в два раза меньше рассчитанных. Уравнение также непригодно для высоких давлений.
Наиболее удовлетворительные результаты дает уравнение Битти – Бриджмена:
pV 2 = RT [ V + В о(1 – b/V)] /(1 – с/VT 3) – А о(1 – а/V), (5.12)
где a, b, c, A, B – эмпирические константы, характерные для каждого газа.
В настоящее время известно более 150 уравнений состояния газов, большинство из которых является эмпирическими, и применимы лишь в определенном интервале температур и давлений. Кроме того, применение таких уравнений приводит к сложным формулам для термодинамических потенциалов, зачастую непригодных для проведения практических расчетов.
Г. Льюис предложил метод расчета термодинамических характеристик реальных газов, основанный на том, что для них формально сохраняются те же соотношения, что и для идеальных газов, но вместо давления вводится новая функция f – термодинамическая летучесть (или фугитивность), более кратко – летучесть.
Для изобарного потенциала постулируется для моля газа
G º G (Т) + RT ln f. (5.13)
Дополнительно вводится условие, согласно которому по мере уменьшения давления величина летучести приближается к величине давления:
lim f / р ® 1 при р ® 0. (5.14)
Из (5.13) следует, что для изотермического процесса
D G = G 2 – G 1 = RT ln(f 2/ f 1). (5.15)
Значения f нужно найти для каждого реального газа при различных давлениях и температурах. Основой для вычисления летучести являются уравнения (5.13) и (5.14).
Продифференцируем уравнение (5.13) по давлению при T = const:
(¶ G /¶ р) Т = RT (¶ln f /¶ р) Т. (5.16)
Так как (¶ G /¶ р) Т = V, то из уравнения (5.16) получим:
d ln f =  dp. (5.17)
dp. (5.17)
Интегрирование в пределах между состояниями 1 и 2 дает:
 . (5.18)
. (5.18)
Для аналитического решения этого уравнения необходимо знать зависимость объема реального газа от давления (например, по уравнению Ван-дер-Ваальса), и подставить ее в подинтегральное выражение.
Более точным является метод, основанный на графическом нахождении интеграла. Для этого экспериментально определяют объем, занимаемый молем газа при разных давлениях при заданной температуре и строят график зависимости V от p. Величину интеграла определяют как площадь под полученной кривой.
Другой способ заключатся в том, что вначале находят объемную поправку, характеризующую степень отклонения объема реального газа от вычисленного по уравнению Менделеева – Клапейрона:
a =  – V. (5.19)
– V. (5.19)
Тогда уравнение (5.18) можно записать в виде
 . (5.20)
. (5.20)
При р 1® 0 летучесть f 1® р 1 и, отбрасывая индекс 2, получаем:
ln f = ln p –  . (5.21)
. (5.21)
 Рис.5.2. Зависимость объемной поправки от давления (Т 1< Т 2< Т 3< Т 4) Рис.5.2. Зависимость объемной поправки от давления (Т 1< Т 2< Т 3< Т 4) |
Для определения f строят график зависимости a = j (р) (рис. 5.1) и графическим интегрированием находят второй член справа уравнения (5.21) при разных величинах давлений.
Летучесть при больших давлениях и низких температурах сильно отличается от давления, например, при T =273 К и p =1200 атм f СО = 2663, а для азота при T = 198 К и p =6000 атм f = 2•106.
Летучесть можно определить как давление, которое должен производить идеальный газ, чтобы оказывать такое же действие, как и реальный газ. С приближением реального газа к идеальному состоянию f по величине приближается к p, и для идеального газа обе величины становятся одинаковыми. Отношение f/p называют коэффициентом летучести или коэффициентом активности:
g = f/р. (5.22)
При малых величинах a и малых давлениях можно считать a постоянной, тогда
ln f = ln p –  . (5.23)
. (5.23)
и
 . (5.24)
. (5.24)
Показательную функцию можно разложить в ряд, ограничившись двумя членами разложения, в результате чего получим:
 и f =
и f =  , (5.25)
, (5.25)
где p ид – давление, которое имел бы идеальный газ, если бы он занимал тот же объем V, что и реальный газ при заданной температуре.
Описанные методы расчета летучести требуют экспериментального определения объема газа при разных давлениях и температурах. Во многих случаях такие данные отсутствуют, и тогда можно воспользоваться обобщенным методом расчета, основанным на принципе соответственных состояний. Если выражать свойства газов не через p, V, T, а с помощью безразмерных единиц – приведенных параметров – приведенного давления p = р/р кр, приведенного объема j = V/V кр, и приведенной температуры t = Т/Т кр, то можно получить приведенное уравнение состояния f (p, j, t) =0, в которое не входит ни одна из величин, характеризующих данное вещество. Согласно принципу соответственных состояний, при одинаковых приведенных параметрах различные реальные газы имеют ряд одинаковых свойств, в том числе и коэффициенты летучести.
 Рис. 5.3. Схематическое изображение зависимости коэффициентов летучести от приведенного давления при разных приведенных температурах Рис. 5.3. Схематическое изображение зависимости коэффициентов летучести от приведенного давления при разных приведенных температурах |
Таким образом, достаточно получить данные о коэффициентах летучести одного газа при различных приведенных давлениях и температурах и эти данные можно использовать для расчетов летучести других газов. Однако, следует отметить, что принцип соответственных состояний является приближенным и строго не выполняется.
Стандартное состояние
Изобарный потенциал реального газа выражается уравнением (5.13), в котором G (T) – постоянная интегрирования, абсолютную величину которой определить невозможно. Итак, нужно ввести некоторое стандартное состояниекак начало отсчета G. Поскольку при р ® 0 летучесть реального газа f ® р, то за стандартное состояние можно было бы взять этот нижний уровень. Но такой выбор практически неудобен, так как при р ® 0 изменение потенциала, по уравнению (5.15), D G ® ¥.
Поэтому за стандартное состояние индивидуального газа при заданной температуре принимается гипотетическое состояние идеального газа, летучесть которого равняется единице, а энтальпия равняется энтальпии реального газа при этой же температуре и нулевом давлении. За единицу давления принимается 1 бар = 1,000.105 Па.
Переход газа из любого состояния в стандартное схематически изображен на рис. 5.4. Если реальный газ находится при данной температуре под давлением р (точка а), проведем изотермическое расширение до бесконечно малого давления (р ® 0), а потом сжатие по изотерме идеального газа к р = 1 (точка о). Это и будет стандартное состояние, в котором f о = р = 1.
Для идеального газа всегда f = р и f o = 1 при любой температуре. Для реального газа при р = 1 его состояние мало отличается от стандартного, то есть можно считать, что практически эти состояния совпадают. Итак, уравнения (5.13), с учетом стандартного состояния, можно записать:
G – G o(Т) = RT ln f / f o = RT ln a. (5.26)
 Рис. 5.4. Переход в стандартное состояние (1 – изотерма реального газа, 2 – изотерма идеального газа) Рис. 5.4. Переход в стандартное состояние (1 – изотерма реального газа, 2 – изотерма идеального газа) |
Отношения летучести в данном состоянии к f o называется активностьюa = f/f o. Поскольку для газов f o =1, то для них f = а.
Для конденсированных фаз (индивидуальной жидкости или твердого тела) в качестве стандартного выбирается реальное состояние вещества при р = 1 при заданной температуре. Жидкость или твердое тело находятся в равновесии с насыщенным паром; таким образом, можно определять свойства коденсированной фазы через свойства насыщенного пара. Влияние давления на летучесть такой фазы описывается уравнением (5.18). Поскольку сжимаемость жидкости или твердого тела очень мала, то практически можно считать, что V = const. Тогда
 . (5.27)
. (5.27)
Зависимость летучести от температуры
Продифференцируем уравнение (5.26) по температуре при р = const, принимая во внимание, что летучесть зависит от температуры:
 (5.28)
(5.28)
или в соответствии с уравнением (5.26):
 . (5.29)
. (5.29)
Поскольку G = H – TS, а (¶ G /¶ T) p = – S (уравн. 4.32), то G = H + T (¶ G /¶ T) p и
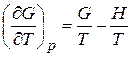 и
и  (5.30)
(5.30)
В стандартном состоянии при любой температуре f o =1 и (¶ln f o/¶ T) p = 0. Подстановка этого значения и уравнения (5.30) в уравнение (5.29) дает:
 , (5.31)
, (5.31)
где Н о и Н – энтальпии газа в стандартном и реальном состояниях, а их различие определяет изменение энтальпии при изотермическом изменении давления от заданного к стандартному.
Если предположить, что это различие не зависит от температуры, то интегрирование уравнения (5.31) дает:
 . (5.32)
. (5.32)
Изменение энтальпии можно рассматривать как затраты энергии на преодоление сил взаимодействия при переходе от реального к идеальному состоянию.
Фазовые превращения. Уравнение Клаузиуса – Клапейрона.
Индивидуальное вещество может находиться в газообразном, жидком или твердом (в различных кристаллических модификациях) состояниях. В равновесной системе, состоящей из нескольких фаз чистого вещества, возможны переходы из одной фазы в другую – плавление, испарение, сублимация, переход из одной кристаллической модификации в другую. Эти процессы характеризуются определенным тепловым эффектом, скачкообразным изменением объема и энтропии при непрерывности изобарного потенциала. Их называют фазовыми переходамипервого рода, или агрегатными превращениями.
Рассмотрим равновесный переход одного моля вещества из одной фазы 1 в другую 2 при постоянных давлении и температуре. В таком процессе производится только работа расширения и изменение внутренней энергии будет равно
U 2 – U 1 = Т (S 2 – S 1) – р (V 2 – V 1), (5.33)
или
U 2 – TS 2 + pV 2 = U 1 – TS 1 + pV 1. (5.34)
Левая и правая суммы равенства равны, по определению, изобарным потенциалам G 2 и G 1 моля вещества в сосуществующих фазах, т.е. изобарные потенциалы единицы массы вещества в двух фазах, находящихся в равновесии, равны между собой:
G 2 = G 1. (5.35)
Изменение температуры (или давления) приводит к нарушению равновесия, и новое равновесие устанавливается при другом давлении (или температуре). При этом изобарные потенциалы фаз изменяются:
dG 1 = – S 1 dT + V 1 dp, (5.31)
dG 2 = – S 2 dT + V 2 dp. (5.32)
Но в новых условиях остаются справедливыми условия равновесия (5.35) и
G 2 + dG 2 = G 1 + dG 1, т.е. dG 2 = dG 1 и после вычитания уравнений получим
(V 2 – V 1) dp = (S 2 – S 1) dT, (5.38)
или
 . (5.39)
. (5.39)
Так как фазовые превращения представляют собой равновесный изотермический процесс, то изменение энтропии
D S = S 2 – S 1 =  , (5.40)
, (5.40)
где D Н ф.п. – изменение энтальпии при фазовом переходе (теплота фазового перехода). После подстановки уравнения (5.40) в (5.39) получим:
 . (5.41)
. (5.41)
Уравнение (5.41) называется уравнением Клаузиуса – Клапейрона и приложимо ко всем агрегатным превращениям индивидуальных веществ.
Рассмотрим некоторые частные случаи использования уравнения Клаузиуса – Клапейрона для фазовых переходов первого рода.
Плавление. В процессе плавления происходит поглощение тепла, т.е. всегда D Н пл > 0. В подавляющем большинстве случаев мольный объем жидкости больше мольного объема твердого тела, т.е. V 2 > V 1, и для процессов плавления в соответствии с уравнением (5.41) dp/dT > 0. Это значит, что при повышении давления температура плавления веществ обычно увеличивается. Исключением является вода (плотность льда меньше плотности жидкой воды), висмут, галий и некоторые другие вещества, для которых V 2 < V 1, поэтому для них dp/dT < 0 и их температура плавления падает при повышении давления.
Испарение. Теплота испарения, как и теплота плавления, всегда положительна. Объем газа (пара) также всегда больше объема жидкости, поэтому всегда dp/dT > 0, т.е. при повышении внешнего давления температура кипения жидкости повышается или при повышении температуры давление насыщенного пара над жидкостью возрастает.
При температурах, далеких от критических, объем газа во много раз больше объема жидкости, поэтому последним можно пренебречь и считать в уравнении (5.41) V 2 – V 1 = V газ. В этих же условиях пар можно рассматривать как идеальный газ, объем которого V = RT/p. Обозначим теплоту испарения D Н = l, и после соответствующих подстановок в (6.9) получим:
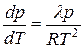 , (5.42)
, (5.42)
или после разделения переменных
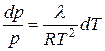 . (5.43)
. (5.43)
Уравнение (5.43) является частным случаем уравнения Клаузиуса – Клапейрона и выражает зависимость давления насыщенного пара над жидкостью от температуры. Если считать, что в небольшом температурном интервале теплота испарения не зависит от температуры, то интегрирование этого уравнения дает:
ln p = –  + const, (5.44)
+ const, (5.44)
т.е. логарифм давления насыщенного пара является линейной функцией обратной величины температуры. Интегрирование уравнения (6.5) в пределах от p 1 до p 2 и от T 1 до T 2 дает
 . (5.45)
. (5.45)
 Рис. 5.5. Зависимость ln p от 1/ Т для насыщенного пара Рис. 5.5. Зависимость ln p от 1/ Т для насыщенного пара |
Теплоты испарения жидкостей зависят от температуры, особенно сильно вблизи критической температуры, где l = 0, поэтому последние два уравнения являются приближенными. Для точных вычислений необходимо учесть температурную зависимость l по уравнению Кирхгоффа, используя данные о теплоемкостях жидкости и пара.
Теплоты испарения жидкостей связаны с их нормальными температурами кипения. По правилу Трутона, мольные энтропии испарения различных жидкостей при нормальных температурах кипения одинаковы:
D S вип =  = 80 – 90 Дж×моль–1×К–1. (5.46)
= 80 – 90 Дж×моль–1×К–1. (5.46)
Правило Трутона приближенно выполняется для углеводородов и их производных, эфиров и других классов неполярных веществ, но не выполняется для полярных и ассоциированных жидкостей (вода, спирты и т.п.).
Более точным является правило Гильдебранда, согласно которому энтропии испарения жидкостей равны между собой при температурах, для которых мольные объемы насыщенного пара одинаковы. При этом изменение энтропии D S составляет 80 – 90 Дж моль–1.К при V = 49,5 л/моль.
Кроме рассмотренных, существуют также фазовые переходы второго рода, для которых характерно не только равенство изобарных потенциалов, но и равенство объемов и энтропий сосуществующих фаз:
D G = 0; D V = 0; D S = 0. (5.47)
Такие превращения не сопровождаются тепловым эффектом, но характеризуются изменением теплоемкости, коэффициента теплового расширения, сжимаемости. К ним относятся переходы ферромагнитных тел в парамагнитные при температуре, называемой точкой Кюри, переходы металлов в сверхпроводящее состояние, превращение гелия I в гелий II и т.п.
Как уже отмечалось, индивидуальное вещество может существовать в нескольких агрегатных состояниях, т. е. образовывать несколько фаз. Например, вода, кроме жидкой и газообразной фаз, образует еще шесть модификаций льда, твердая сера имеет ромбическую и моноклиническую модификации и т.п. Однако, максимальное число равновесно сосуществующих фаз обычно меньше общего числа возможных фаз. Это максимальное число можно определить по правилу фаз Гиббса (4.57):
f = k + 2 – s. (5.48)
Так как f максимально при s = 0, а k = 1, то максимальная величина f = 3, т.е. одновременно в однокомпонентной гетерогенной системе может находиться в равновесии не более трех фаз.
В общем случае нам не известны уравнения состояния отдельных фаз, поэтому зависимость между переменными, определяющими состояние системы, устанавливается при экспериментальном измерении температуры, давления и концентраций или объемов равновесных систем.
Эти данные используют для построения диаграмм состояния, которые являются графическим выражением этих зависимостей.
В случае однокомпонентной системы в уравнение состояния входит три переменных: температура T, давление p и концентрация c; или T, p и мольный объем V. Для графического изображения связи между T, p и V необходимо использовать систему координат из трех взаимно перпендикулярных осей, каждая из которых соответствует одной из переменных. Любое состояние системы отображается в такой системе координат одной точкой, которая называется фигуративной точкой.
Совокупность этих точек дает диаграмму (рис. 5.6), состоящую из нескольких поверхностей, определенным образом расположенных в пространстве. Точки вне этих поверхностей не имеют физического смысла, так как любая фаза при заданных давлении и температуре имеет строго определенный объем, т.е. каждому сочетанию T и p соответствует единственно возможное значение V для данной фазы. Исключением являются точки на поверхностях, соединяющих границы двух фазовых поверхностей. В этом случае фигуративная точка описывает средний мольный объем вещества во всей системе.
 Рис. 5.6. Схематическое изображение объемной диаграммы состояния однокомпонентной системи и ее проекций на плоскости Рис. 5.6. Схематическое изображение объемной диаграммы состояния однокомпонентной системи и ее проекций на плоскости |
Подобные объемные диаграммы, позволяющие проследить за изменением всех переменных в уравнении состояния, называют полными диаграммами состояния. Построение полных диаграмм трудоемко, а работать с ними неудобно, поэтому на практике обычно используют проекции полной диаграммы на одну из плоскостей, проходящих через оси координат. Наиболее употребительными параметрами, определяющими условия существования системы, являются T и p, они хорошо поддаются измерению и регулированию. Поэтому чаще всего пользуются проекциями на плоскость T – p. Откладывая значения этих двух переменных по двум осям прямоугольной системы координат, получают двумерную (плоскую) диаграмму. Каждая фигуративная точка на плоскости выражает условия (T и p), при которых находится система. Это позволяет разбить всю плоскую диаграмму на отдельные области, каждая из которых охватывает все возможные сочетания Т и р, отвечающие равновесному существованию определенной фазы (рис. 5.6). На рисунке область газ отвечает условиям существования газообразной фазы, область жидк. – жидкой фазы и область тв. – твердой фазы. Пограничные линии Ок, Оа, Оо принадлежат обеим соприкасающимся областям и каждая точка на этих линиях может отвечать как совместному сосуществованию обеих фаз, так и наличию только одной из фаз. Это обусловлено тем, что любой фазовый переход при постоянных T и p сопровождается изменением энтальпии. Поэтому, например, в точке a жидкость и кристаллы сосуществуют лишь в том случае, когда энтальпия системы выше энтальпии твердого состояния, но ниже энтальпии жидкости, т.е. когда фазовый переход еще не завершен. Если же фазовый переход еще не начинался или уже завершен, система представляет одну фазу.
В тройной точке O также возможно сосуществование в равновесии всех трех фаз, но возможно также сосуществование двух каких-либо фаз или наличие только одной фазы.
В соответствии с правилом фаз в каждой из областей система имеет две степени свободы, т. е. можно произвольно менять давление и температуру без изменения фазового состава системы. Если фигуративная точка системы лежит на какой-либо из разделяющих линий и соответствует равновесному сосуществованию двух фаз, то система является моновариантной, т.е. две фазы могут оставаться в равновесии только при произвольном изменении или температуры или давления. При изменении T давление должно приобретать строго определенное значение и наоборот, т.е. фигуративная точка системы должна перемещаться по разделяющей линии.
Три фазы могут находиться в равновесии только при строго определенных значениях T и p, соответствующих тройной точке O, и в этих условиях система является инвариантной (s = 0).
Разделяющие кривые на диаграмме отражают условия агрегатных превращений вещества и описываются уравнениями Клаузиуса – Клапейронадля соответствующих переходов. Поэтому кривая Oa, соответствующая процессу плавления вещества, для воды, висмута, галия будет иметь наклон влево в отличие от большинства других веществ, для которых мольный объем жидкости больше объема твердого вещества.
Кривая равновесия жидкость – газ Oк заканчивается в критической точке к. Выше критической температуры вещество может существовать только в газообразном состоянии при любых давлениях.
Энантиотропные и монотропные превращения
Как уже говорилось выше, вещества в твердом состоянии могут иметь разные кристаллические модификации. Поэтому на диаграммах состояния должны появляться области, соответствующие существованию этих модификаций. На рис. 5.7 схематически показана диаграмма состояния серы.
 Рис. 5.7. Схематическая диаграмма состояния серы Рис. 5.7. Схематическая диаграмма состояния серы |
Сплошные линии делят диаграмму на четыре области, которые отвечают условиям равновесного существования пара, жидкости и двух кристаллических модификаций – серы ромбической и серы моноклинной. Линии отвечают условиям, при которых возможно равновесное сосуществование двух соответствующих фаз. В точках A, B и C в термодинамическом равновесии находятся три фазы. Кроме того, существует еще одна тройная точка о, в которой могут сосуществовать перегретая ромбическая сера, переохлажденная жидкая сера и пар, пересыщенный относительно пара, равновесного с моноклинной серой.
Химические потенциалы трех фаз при температуре и давлении, отвечающих точке о, одинаковы. Благодаря этому три термодинамически неустойчивые фазы образуют метастабильную систему, т.е. систему, находящуюся в состоянии относительной устойчивости. Метастабильностьзаключается в том, что ни одна из трех фаз не стремится перейти в другую, но при длительной выдержке или внесении кристаллов моноклинной серы все три фазы переходят в моноклинную серу, которая является единственной термодинамически устойчивой фазой в этих условиях.
Метастабильные тройные точки могут давать только те вещества, которые образуют несколько кристаллических модификаций. Метастабильными являются также равновесия между двумя фазами, которым соответствуют кривые оA, оB и оC.
Если одна кристаллическая модификация при повышении температуры должна перейти в другую, то возможен некоторый перегрев выше температуры устойчивого равновесия. Это объясняется тем, что переход из одной кристаллической модификации в другую затруднен.
Перегретая модификация должна быть некоторое время выдержана при достигнутой температуре для того, чтобы произошла перестройка кристаллической решетки. Если же кристаллы достигли температуры плавления, то дальнейший подвод тепла приводит к немедленному разрушению решетки.
Химические потенциалы серы ромбической и серы моноклинной могут быть выражены через давления равновесных с ними паров:
m р = m о + RT ln p р и m м = m о + RT ln p м. (5.49)
Так как m о имеет одно и то же значение, следовательно, химический потенциал будет больше у той модификации серы, давление насыщенных паров которой выше.
Кривая давления пара ромбической серы EAо и кривая давления пара моноклинной серы DAC пересекаются в точке A, соответствующей температуре 95,5 С и лежащей ниже кривой давления пара над жидкостью кCо. При температурах ниже 95,5 С давление пара моноклинной серы выше давления пара ромбической серы. Поэтому при температуре, например, Т 1 возможен самопроизвольный переход Sм ® Sр.
При температурах выше 95,5 С, например, Т 2 давление пара выше у серы ромбической и превращение должно идти в обратном направлении: Sм ® Sр. Взаимные превращения двух кристаллических модификаций, которые могут протекать самопроизвольно в прямом и в обратном направлениях в зависимости от условий, называются энантиотропными превращениями.
 Рис. 5.8. Схема зависимости давления пара от температуры при монотропних превращениях Рис. 5.8. Схема зависимости давления пара от температуры при монотропних превращениях |
 Рис. 5.9. Диаграмма состояния фосфора (схема) Рис. 5.9. Диаграмма состояния фосфора (схема) |
Бывают случаи, когда кривые зависимости давления паров от температуры для двух кристаллических модификаций должны пересечься выше кривой давления пара над жидкостью (рис. 5.8). Поскольку кристаллы нельзя перегреть выше их точки плавления, обе модификации плавятся соответственно в точках А и В. Диаграмма показывает, что давление пара модификации I во всей области существования кристаллов выше давления пара модификации II, а следовательно, самопроизвольные превращения возможны лишь в направлении I ® II, например, после того, как из жидкой фазы, переохлажденной до температуры T 1, выделится модификация I. Точка пересечения O не может получена экспериментально, а находится графически путем продолжения опытных кривых AC и BD.
Взаимные превращения кристаллических фаз, которые могут протекать самопроизвольно лишь в одном направлении, называются монотропными.
Примером диаграммы, описывающей монотропные переходы, может служить диаграмма состояния фосфора, схематически приведенная на рис. 5.9.
Кривые давления паров неустойчивых фаз обозначены на диаграмме пунктиром. Точка o соответствует устойчивому сосуществованию жидкого фосфора, твердого фиолетового фосфора и парообразного фосфора. Точки E и D соответствуют неустойчивому сосуществованию трех фаз; точка D – двух модификаций фосфора белого (I b II) и пара; точка E – белого фосфора I, переохлажденного жидкого фосфора и пара. Вся область слева от кривых BOA соответствует устойчивому твердому фиолетовому фосфору.
Диаграмма состояния фосфора представляет собой пример системы, в которой могут происходить как монотропные превращения (фосфора белого I в фосфор фиолетовый), так и энантиотропные превращения (фосфора белого I в фосфор белый II, хотя оба они являются неустойчивыми модификациями по сравнению с фиолетовым фосфором).

