2020-01-15
2020-01-15 295
2952.2.2. В базе данных Pfam представлена доменная структура EP4-рецептора и приводятся выравнивания для отдельных доменов. Основной домен EP4-рецептора (аминокислоты с 44 по 329) носит название 7tm_1 и имеется у всех представителей семейства родопсиновых рецепторов. Состоит он из 7 трансмембранных α-спиралей, соединенных внемембранными участками.
Приводится выравнивание 7tm_1-домена EP4-рецептора человека с 7tm_1-доменом различных представителей семейства родопсиновых рецепторов.
Также выделены
1) сигнальный участок — аминокислоты с 1 по 43, находящиеся вне клетки, и
2) участок, состоящий из сравнительно немногих видов аминокислот (low complexity region), состоящий из аминокислот с 389 по 405.
Выравнивается каждый из этих двух участков EP4-рецептора человека с соответствующими участками EP4-рецепторов кролика, крысы и мыши, а также четырех неаннотированных белков (из базы данных EMBL). 3 из них являются EP4-рецепторами собаки, шимпанзе и макаки, а 4-ый — это неизвестный белок мыши, очень похожий на белок EP4-рецептора. Это приводит к тому же выводу, что и в предыдущем пункте: серьезное сходство последовательностей С- и N-конца проявляется только у рецепторов одного подтипа, принадлежащих разным животным.
2.2.3. Для выявления возможного сходства аминокислотных последовательностей тех участков, которые важны для связывания рецептора с G-белком, выполнено множественное выравнивание (с последующим ручным редактированием) последовательности EP4 - рецептора человека с последовательностями других представителей семейства G-белок-связывающих рецепторов, которые, как и EP4-рецептор, связываются с Gs-белком. Взяты последовательности рецепторов к фолликулостимулирующему гормону, лютеинизирующему гормону, тиреотропному гормону, меланокортину МС3, вазопрессину М2, серотонину 5НТ4, аденозину А2А; b1-рецептора к адреналину, Н2-рецептора к гистамину и D1-рецептора к дофамину. Все эти белки являются белками человека.
Консервативными являются 9 аминокислотных остатков (рис. 6). Находятся они в трансмембранных участках белков. Значительного сходства аминокислотных последовательностей тех участков, которые важны для связывания с G-белком, не наблюдается. Это значит, что основную роль при связывании с G-белком играет пространственная структура соответствующих участков, а не непосредственно аминокислотная последовательность.
2.2.4. Выполнено парное выравнивание белковых последовательностей EP4 - рецептора человека и родопсина быка — единственного представителя класса А G-белок-связывающих рецепторов, для которого есть данные рентгеноструктурного анализа.
В данных двух белковых последовательностях совпадают 47 аминокислотных остатков. Они не собраны в крупные группы, а рассредоточены по всей длине белков. То есть сходство последовательностей не настолько велико, чтобы на это можно было серьезно опираться при построении трехмерной модели EP4-рецептора. К тому же, трехмерная структура известна только для неактивной формы родопсина, в то время как для фармацевтических целей важна активная форма белка. Также необходимо учесть, что структура внеклеточных петель родопсина, возможно, значительно изменена: она определялась прежде всего контактами в кристалле [8].
2.2.5. Сравнение полученных результатов с базой данных по G-белок-связывающим рецепторам (GPCRDB).
В GPCRDB приведены множественные выравнивания для различных групп G-белок-связывающих рецепторов. Выравнивание представителей класса А содержит 1122 белковых последовательностей (включая неаннотированные — из базы данных EMBL). Стоит отметить, что представлены не целые последовательности, а только фрагменты с наибольшим сходством — в основном, трансмембранные, так как вероятность мутации в трансмембранной части ниже, чем во внутри- или внеклеточной части белка.
Авторы пишут о необходимости ручного выравнивания с учетом всех известных экспериментальных данных, если речь идет о большом числе белковых последовательностей [9].
Также в GPCRDB представлены трехмерные модели многих белков, в том числе и EP4-рецептора человека. Множественные выравнивания и трехмерные модели созданы с помощью программы WHAT IF. Она позволяет учесть многочисленные данные, в том числе и трехмерную структуру родопсина, но тем не менее полученный результат не может считаться достаточно точным для фармацевтических целей (об этом предупреждают сами авторы моделей). Пользование программой WHAT IF платное.
Кроме того, в GPCRDB приводятся описания и результаты других методик исследования, способных выявлять более сложные взаимосвязи и закономерности, чем просто анализ расположения консервативных позиций в множественном выравнивании. Далее приводится описание этих методик (в соответствии со статьей [9]).
Для анализа функциональной значимости отдельных аминокислотных позиций выравнивания используется расчет для каждой позиции двух величин: энтропии и вариабельности. Энтропия в позиции p (S p) определяется так:
20
S p = - ∑ f pi * ln (f pi),
i =1
где i пробегает все 20 видов аминокислот, а f pi — это относительная частота аминокислотного остатка i в позиции p выравнивания. Значение S p максимально,
если f p1 = f p2 =... = f p20; минимально (то есть равно нулю), если в позиции встречается только 1 вид аминокислотных остатков. Таким образом, S p можно назвать мерой информации, содержащейся в позиции p выравнивания.
Вариабельность в позиции p (V p) определяется как число разных видов аминокислот, встречающихся в позиции p не менее чем в 0,5 % последовательностей. Можно сказать, что V p является мерой свободы, или хаоса, в позиции p выравнивания.
Все аминокислотные позиции разделяются на 5 блоков в зависимости от своей энтропии и вариабельности (не указано, какие значения приняты пороговыми):
1) блок 11 содержит позиции с низкой энтропией и низкой вариабельностью, которые формируют главный функциональный сайт (для G-белок-связывающих рецепторов —участок связывания с G-белком) или играют важную структурную роль (например, образуют дисульфидные мостики);
2) блок 12 содержит аминокислотные остатки из внутренней части (“сердцевины”) белка; пространственно они расположены вокруг главного функционального сайта;
3) блок 22 содержит по большей части аминокислотные остатки из “сердцевины” белка, расположенные дальше от главного функционального сайта; они играют структурную роль и, возможно, участвуют в передаче сигнала от лиганда к главному функциональному сайту;
4) блок 23 содержит большинство остатков, участвующих во взаимодействиях с лигандом; они могут быть расположены как в “сердцевине” белка, так и на его поверхности;
5) блок 33 содержит в основном позиции, расположенные на поверхности белка и участвующие во взаимодействии с лигандом, но не в передаче сигнала к главному функциональному сайту. Здесь также находится большинство “непокорных” (recalcitrant) позиций — позиций, для которых выравнивание является сомнительным.
Сигнал передается по такому пути: лиганд —> блок 23 —> блок 22 —> блок 12 —> блок 11 —> взаимодействие с G-белком.
Для исследования белковых семейств, состоящих из очень большого числа белков, предлагается анализ коррелированных мутаций (correlated mutation analysis, или CMA). Он используется для выяснения роли отдельных аминокислотных остатков в структуре и функциональной активности белка. СМА позволяет выявить группы (“сетки”) аминокислотных остатков, мутации которых происходят взаимосвязанно (а значит, для поддержания функциональной активности белка важно не столько положение отдельных аминокислотных остатков, сколько образуемая группой остатков структура в целом).
Основными входными данными для СМА являются множественные выравнивания, поэтому они делаются с учетом всех имеющихся экспериментальных данных и доступной на данный момент информации о трехмерной структуре белков. В процессе анализа учитываются только те части, пространственная укладка которых является общей для всех белков в данном выравнивании. Участки, где есть вставки и делеции, не анализируются.
В зависимости от целей в СМА применяются различные матрицы:
· для изучения структурных аспектов используются матрицы Dayhof (Dayhof-style residue similarity matrices);
· для изучения функциональных аспектов используется единичная матрица (identity matrix for residue similarities) — аминокислотные остатки или одинаковы, или различны; никаких промежуточных значений нет.
Степень сходства (correlation score) между позициями p и q вычисляется по следующей формуле:
n -1 n
С(p, q) = W(i, j) * ∑ ∑ δ(ip, iq, jp, jq),
p =1 q=p +1
где n — число белковых последовательностей в выравнивании. Пара чисел p и q пробегает все пары позиций в последовательностях i и j. ip, iq, jp и jq образуют две пары позиций (p, q) в двух последовательностях — i и j, соответственно. Функция δ такова, что
· δ(ip, iq, jp, jq) = 1, если ip = jp и iq = jq или ip ≠ jp и iq ≠ jq;
· δ(ip, iq, jp, jq) = 0, если ip = jp и iq ≠ jq или ip ≠ jp и iq = jq.
W(i, j) — вес (weight factor) пары последовательностей i и j, который вычисляется так:
n -1 n
W(i, j) = wi * wj / ∑ ∑ wk * wl,
k =1 l=k +1
где wi и wj — веса последовательностей i и j.
Следующий пример (таблицы 2 и 3 с пояснениями) иллюстрирует первую формулу. Для простоты все веса приняты равными 1,0.
Таблица 2.
| № последовательности | № позиции | ||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| 1 | A | S | P | W | L | R | L |
| 2 | A | S | P | W | I | D | L |
| 3 | T | T | P | W | V | K | G |
| 4 | T | G | P | W | M | E | G |
Взяты 4 произвольные последовательности, из 7 аминокислотных остатков каждая.
Степень сходства С(1, 2) рассчитывается так:
n -1 n
С(1, 2) = (2 / n *(n -1)) * ∑ ∑ δ(1 p, 1 q, 2 p, 2 q) = 1/6 * (δ(ASAS) + δ(ASTT) + δ(ASTG) +
p =1 q=p +1
+ δ(ASTT) + δ(ASTG) + δ(TTTG)) = 1/6 * (1+1+1+1+1+0) = 0,84
Остальные степени сходства рассчитываются аналогично; их значения для указанных 10 пар позиций приведены в таблице 3.
Таблица 3.
| p, q | 1, 2 | 1, 3 | 1, 4 | 1, 5 | 1, 6 | 1, 7 | 2, 3 | 3, 4 | 5, 6 | 6, 7 |
| С(p, q) | 0,84 | 0,33 | 0,33 | 0,67 | 0,67 | 1,00 | 0,16 | 1,00 | 1,00 | 0,67 |
Если все попарные степени сходства в некоторой группе позиций превышают величину, принятую за точку отсчета (обычно 0,7 – 0,9), то эта группа называется “сетью” (network).
Позиции, в которых наблюдается консервативность у некоторых подсемейств внутри анализируемого семейства белков, называются “непокорными” (recalcitrant). Они не используются ни в одном из видов СМА. Эти позиции часто демонстрируют высокие степени сходства, но функционально важными не являются. В G-белок-связывающих рецепторах они часты в близких к трансмембранным спиралям внемембранных участках.
Высокая степень сходства каких-либо двух позиций может свидетельствовать об одной из трех вещей:
1) обе данные позиции полностью консервативны;
2) обе позиции вариабельны, причем имеют одинаковые вариабельность и энтропию;
3) мутации в этих позициях скоррелированы (аминокислотные остатки или вместе остаются консервативными, или вместе заменяются).
Пункт 1 — это частный случай пункта 3; а если число последовательностей в выравнивании очень велико, то и пункт 2 становится частным случаем пункта 3.
С помощью СМА в G-белок-связывающих рецепторах выделены 3 “сетки” (network):
· 1-ая состоит из наиболее консервативных аминокислотных остатков, участвующих в связывании G-белка;
· 2-ая состоит из остатков, участвующих в связывании лиганда;
· 3-ая состоит из остатков, участвующих в связывании и активации G-белка.
То есть “сетки” состоят из аминокислотных остатков, важных для связывания или с лигандом, или с G-белком. А “сеток”, связанных с передачей сигнала, нет. Это можно объяснить или отсутствием строгого давления эволюции на аминокислотные остатки, расположенные в центральной части белка; или тем, что важна структура центральной части в целом, и СМА не может выявить столь сложные взаимосвязи.
Выводы.
PG-рецепторы — это трансмембранные белки, поэтому количество гидрофильных аминокислотных остатков в PG-рецепторах меньше среднестатистического для белков человека, а количество гидрофобных — больше.
PG-рецепторы являются близкими гомологами друг для друга, при этом их аминокислотные последовательности заметно отличаются от аминокислотных последовательностей прочих белков. Максимальное сходство аминокислотных последовательностей наблюдается у представителей одного типа (или, если есть, подтипа) PG-рецепторов, принадлежащих разным организмам. Это позволяет предположить, что создание высокоселективных агонистов или антагонистов к какому-либо одному виду PG-рецепторов является осуществимой задачей. Также можно предположить, что эти агонисты/антагонисты будут одинаково действенны для данного вида рецептора, вне зависимости от вида животного. Это значит, что при разработке лекарств для человека можно будет проводить исследования на других животных.
Более далекие гомологи PG-рецепторов принадлежат, как и сами PG-рецепторы, к семейству А G-белок-связывающих рецепторов. Сходство аминокислотных последовательностей в данной группе невелико, и консервативными для 100 представителей этого семейства являются только 6 аминокислотных остатков.
Выполненное автоматически множественное выравнивание большого числа трансмембранных белков требует последующего ручного редактирования. При этом следует опираться на аминокислотные последовательности трансмембранных α-спиралей.
Выявлены мотивы, характерные для всех PG-рецепторов. Они частично совпадают с мотивами, свойственными большинству представителей семейства А G-белок-связывающих рецепторов.
Для всех PG-рецепторов абсолютно консервативными являются 27 аминокислотных остатков; для PG-рецепторов человека — 28 аминокислотных остатков; для ЕP-рецепторов — 40 аминокислотных остатков. На основании этих результатов нельзя сделать каких-либо определенных выводов о пространственной структуре PG-рецепторов. Достоверное моделирование трехмерной структуры PG-рецепторов на основе данных рентгеноструктурного анализа для родопсина также невозможно.
Для Gs-связывающих белков консервативными являются 9 аминокислотных остатков, расположенные преимущественно в трансмембранных участках. Это свидетельствует о том, что для связывания с G-белком важна не столько аминокислотная последовательность соответствующих участков, сколько их пространственная структура.
Все вышесказанное демонстрирует, что исследование трансмембранных белков требует значительно более сложного подхода, чем просто анализ множественных выравниваний, сделанных автоматически. Необходимо редактирование выравниваний с учетом всех имеющихся экспериментальных данных. Для получения из выравниваний информации о функциональной важности участков белка требуется рассмотрение более сложных взаимозависимостей, чем одна только консервативность аминокислот. Одна из возможных методик анализа множественных выравниваний — СМА.
Использованная литература
1. Варфоломеев С. Д. Простагландины – новый тип биологических регуляторов. (1996) Соросовский Образовательный журнал.
2. Breyer R., Bagdassarian C., Myers S., Breyer M. (2001) Prostanoid Receptors: Subtypes and Signaling. Annu. Rev. Pharmacol. Toxicol. 41:661-90.
3. Геннис Р. Биомембраны: Молекулярная структура и функции. М., Мир, 1997.
4. Bockaert J., Pin J.P. (1999) Molecular tinkering of G protein-coupled receptors: an evolutionary success. The EMBO Journal, Vol.18, No.7, pp. 1723-1729
5. Audoly L., Breyer R. (1997) Substitution of Charged Amino Acid Residues in Transmembrane Regions 6 and 7 Affect Ligand Binding and Signal Transduction of the Prostaglandin EP3 Receptor. Molecular Pharmacology, 51:61-68.
6. Audoly L., Breyer R. (1997) The Second Extracellular Loop of the Prostaglandin EP3 Receptor Is an Essential Determinant of Ligand Selectivity. The Journal of Biological Chemistry, Vol. 272, No. 21, Issue of May, pp. 13475-13478.
7. Horn F., Bettler E., Oliveira L., Campagne F., Cohen F., Vriend G. GRCRDB information system for G protein-coupled receptors. (2003) Nucleic Acids Research, Vol. 31, No. 1, pp. 294-297.
8. Oliveira L., Hulsen T., Lutje Hulsik D., Paiva A., Vriend G. Modelling G protein-coupled receptors. GRCRDB, http://www.gpcr.org/7tm/articles.
9. Oliveira L., Paiva A., Vriend G. Correlated Mutation Analyses on Very Large Sequence Families. GRCRDB, http://www.gpcr.org/7tm/articles/2002_3.
10. Oliveira L., Paiva P., Paiva A., Vriend G. Sequence analysis reveals how G protein-coupled receptors transduce the signal to the G protein. GRCRDB, http://www.gpcr.org/7tm/articles/2002_2.



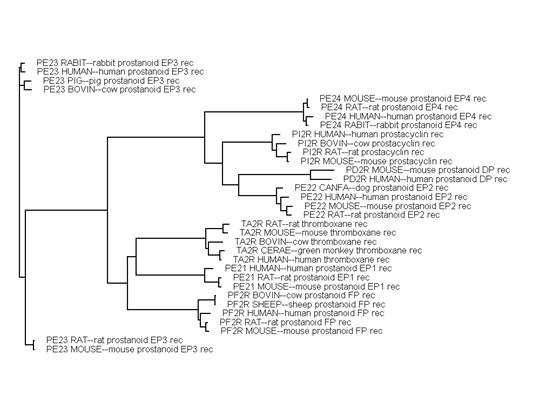
Рис. 4. Филогенетическое древо для простаноидных рецепторов. Корень – EP3-рецептор человека. Получено с помощью программы BLAST (с сервера NCBI) по 34 белкам из базы данных SwissProt.
(“rec” означает “receptor”.)
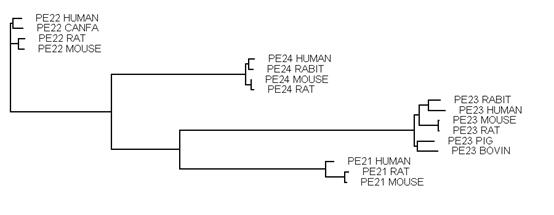 |
Рис. 5. Филогенетическое древо для EP-рецепторов. Корень – EP4-рецептор человека.