2020-05-25
2020-05-25 304
304Министерство науки и высшего образования Российской Федерации
ФГАОУ ВО «УрФУ имени первого Президента России Б.Н. Ельцина»
Химико-технологический институт»
Кафедра иммунохимии
МЕТОДИЧЕСКИЕ УКАЗАНИЯ
для проведения лабораторного занятия по теме
«Электрофорез мышечных белков по Лэммли»
Екатеринбург
2020
Оглавление
Введение. 3
1. Теоретическая часть. 4
1.1. Принцип метода. 4
1.2. Реактивы.. 5
1.3. Применение. 8
1.4. Преимущества и недостатки. 8
1.5. Характеристика разделяемых белков. 9
2. Практическая часть. 10
2.1. Приготовление рабочих растворов. 10
2.2. Ход работы.. 10
2.3. Интерпретация результатов. 12
2.4. Подготовка отчета. 13
3. Контроль знаний. 14
3.1. Контрольные вопросы.. 14
3.2. Лист ответов. 16
Библиографический список. 17
Введение
Электрофорез на протяжении последних десятилетий традиционно занимает важное место при исследовании белков и нуклеиновых кислот. Метод позволяет разделять макромолекулы по размерам, пространственной конфигурации, вторичной структуре и заряду, что в сочетании с простотой и удобством в использовании делает его незаменимым не только для качественного, но и для количественного анализа макромолекул.
Впервые данный подход был применён шведским химиком Арне Тизелиусом в 1930-х годах для разделения белков сыворотки крови, а уже в 1948-м году, эта работа была отмечена нобелевской премией. С тех пор электрофорез успешно применяется для разделения почти всех известных органических и неорганических соединений. Наибольшие успехи метода электрофореза связаны с его способностью разделять заряженные биологические макромолекулы, что нашло широкое применение не только в биологических и биохимических исследованиях, но и в биотехнологии. На протяжении многих лет электрофорез является основным методом разделения белков, оставаясь одним из самых чувствительных методов.
1. Теоретическая часть
1.1. Принцип метода
Электрофорез – это электрокинетическое явление перемещения частиц дисперсной фазы (коллоидных или белковых растворов) в жидкой или газообразной среде под действием внешнего электрического поля.
В растворе белки находятся в виде заряженных частиц. Заряд на поверхности белков возникает в результате диссоциации группировок, находящихся в боковых радикалах аминокислот (карбоксильных, амино-, имидазольных и др. групп), а также при связывании ионов. Так как степень диссоциации группировок зависит от рН раствора, то величина и знак суммарного заряда белковой молекулы зависят от рН среды, а также от ионной силы (интенсивности электрического поля, создаваемого ионами в растворе).
Для каждого белка существует такое значение рН среды (обозначаемое как рI – изоэлектрическая точка), при котором положительные и отрицательные заряды ионизированных групп скомпенсированы, поэтому заряд всей белковой молекулы равен нулю. В буфере с рН, равным рI изучаемого белка, отсутствие заряда на белковой молекуле делает невозможным ее движение в электрическом поле. Из-за разницы в аминокислотном составе разные белки имеют разные значения рI.
При рН ≠ pI молекулы белка приобретают заряд и под действием электрического поля перемещаются к противоположно заряженному электроду – катоду (-) или аноду (+). Например, кислые белки, богатые моноаминодикарбоновыми аминокислотами (аспарагиновая кислота, глутаминовая кислота), в слабощелочном буфере приобретут отрицательный суммарный заряд из-за диссоциации СООН- групп до СОО- и H+ и будут двигаться к аноду. Для электрофоретического разделения оптимально такое значение рН рабочего буфера, которое обусловливает максимальное различие зарядов разных белков, составляющих исходную смесь, а не их максимальный заряд. Обычно электрофорез проводят в среде (буфере) со значением рН, на 3–4 единицы отличающимся от среднего значения рI для белков данного типа. Это позволяет добиться хорошей электрофоретической подвижности (см. ниже) и вместе с тем – сохранить ощутимые различия молекул по заряду. Предпочтительно использовать буфер известной и постоянной ионной силы на основе однозарядных ионов. От рабочего буфера также требуется существенная емкость, так как локальная концентрация белка в образующихся при разделении смеси зонах скопления молекул может оказаться значительной.
При проведении электрофореза электрическое поле создают с помощью источника питания – стабилизированного выпрямителя, способного давать регулируемое напряжение при силе тока в несколько десятков миллиампер (мА). Электрофорез проводят в однородном электрическом поле, то есть поле, напряженность E которого во всех точках одинакова.
Электрический ток пропускают через проводник – буферный раствор. Сопротивление буферного раствора задается двумя факторами: концентрацией в нем свободных ионов и их электрофоретической подвижностью. Электрофоретическая подвижность – скорость движения заряженной молекулы (выражаемой в см/ч) в электрическом поле с напряженностью 1 В/см. Именно различия в электрофоретической подвижности белков, содержащихся в анализируемой смеси, делают возможным разделить эти белки в пространстве (в разных зонах электрофореграммы).
При электрофорезе работа силы трения заряженных компонентов разделяемой смеси о среду приводит к нагреву геля. Кроме того, образование тепла вызывается прохождением электрического тока. В итоге происходит значительное возрастание температуры, которое ухудшает результаты электрофоретического разделения, так как изменяет вязкость и проводимость среды, увеличивает скорость диффузии молекул, способствует испарению летучих компонентов, приводит к денатурации белков. Поэтому при проведении электрофореза следует обеспечить охлаждение системы (например, помещая прибор для электрофореза в холодильную камеру).
1.2. Реактивы
Вертикальный электрофорез белков в денатурирующем полиакриламидном геле – одна из форм зонального электрофореза метод разделения макромолекул, в котором используется неоднородная разделяющая система с полиакриламидным гелем в качестве носителя; пара буферов имеет разный состав и разные значения рН, а носитель состоит из отдельных слоев геля, различающихся по размерам пор.
Полиакриламидный гель (ПААГ) имеет свойства молекулярного сита, чем обеспечивает электрофоретическое разделение белковых смесей не только по заряду, но и по размеру и форме частиц. При электрофорезе в ПААГ крупные молекулы, размеры которых соизмеримы с диаметром пор геля, движутся медленнее, а мелкие молекулы свободно и быстро проходят через поры геля.
ПААГ формируют путем сополимеризации акриламида, создающего линейную «основу», и N,N′-метиленбисакриламида, служащего для поперечных «сшивок» линейных цепей (рис. 1.1). Меняя концентрацию акриламида от 2 до 50 % можно задать определенную пористость геля.

Рис. 1.1. Структурные формулы акриламида и бисакриламида
В результате сополимеризации образуется трехмерная сетка геля, строение фрагмента которой представлено на рис. 1.2. Каждый второй углеродный атом линейной цепи содержит кислотную амидную группу, что обеспечивает гидрофильность полимера. В то же время ПААГ не содержит ионизируемых групп.
Для сополимеризации нужны инициаторы и катализаторы (окислительно-восстановительные системы – источники свободных радикалов).
Персульфат аммония используется в качестве инициатора процесса полимеризации. Тетраметилэтилендиамин используется как катализатор образования ПААГ (рис. 1.3).
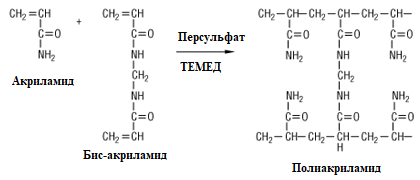
Рис. 1.2. Реакция полимеризации геля
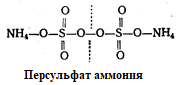
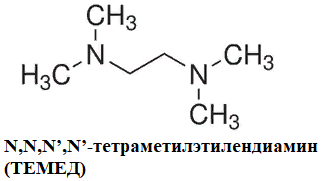
Рис. 1.3. Структурные формулы персульфата аммония и тетраметилэтилендиамина
Гель состоит из двух частей. Концентрирующий гель имеет pH 6,8 и концентрацию полиакриламида от 2 до 12 %. Разделяющий гель имеет рН в районе 8,8 и концентрацию полиакриламида от 5 до 20 %. Выбор плотности геля зависит от молекулярных масс исследуемых белков. Все буферы не содержат неорганических солей, основным переносчиком тока в них является глицин (рис. 1.4).
NH2 —CH2 — COOH
Рис. 1.4. Структурная формула глицина
Глицин входит в состав рабочего буфера для создания нужного значения рН. При рН 6,8 суммарный заряд молекулы глицина близок к нулю. Вследствие этого для переноса определенного заряда (который определяется силой тока в электрофоретической ячейке), отрицательно заряженные комплексы полипептидов с ДСН должны двигаться с большой скоростью. При рН 8,8 глицин приобретает отрицательный заряд, вследствие чего на границе концентрирующего и разделяющего гелей белки резко тормозятся (в переносе одинакового заряда через единицу площади теперь участвует гораздо больше заряженных молекул, следовательно, они двигаются с меньшей скоростью). Результатом этого является концентрирование белков на границе гелей, что очень сильно повышает разрешающую способность метода.
Для разделения белков в соответствии с их молекулярной массой перед началом электрофореза белковую смесь инкубируют в присутствии додецилсульфата натрия (ДСН) (рис. 1.5). Анионый детергент ДСН прочно связывается с большинством белков в соотношении 1,4 мг ДСН /мг белка, при этом суммарный комплекс белок-(ДСН)n приобретает отрицательный заряд, кроме того, ДСН приводит к разрушению нековалентных связей, приводя к денатурации белков.
Благодаря дополнительной обработке белков реагентами, восстанавливающими дисульфидные связи (рис. 1.5) 2-меркаптоэтанолом или дитиотреитолом (ДТТ) происходит дальнейшая денатурация белков и разрушение белок-белковых комплексов. В этом случае единственным фактором, который может повлиять на подвижность белков в полиакриламидном геле является размер белка, а точнее его молекулярная масса. Подвижность белок-(ДСН)n обратно пропорциональна логарифму молекулярной массы: низкомолекулярные комплексы движутся быстрее, чем высокомолекулярные. Это означает, что молекулярная масса белков может быть определена по относительной подвижности белка в геле, а наличие одной полосы в таком геле может являться хорошим критерием чистоты препарата.
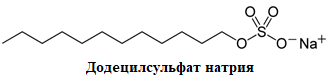
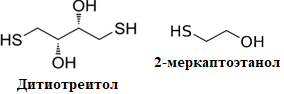
Рис. 1.5. Структурные формулы ДСН, ДТТ и 2-меркаптоэтанол
Трис(гидроксиметил)аминометан (Tris Base) используется в качестве компонента буферного раствора.
В ходе электрофореза зоны растворенных макромолекул остаются невидимыми. Для наблюдения за процессом в исходный препарат добавляют краситель, молекулы которого несут электрический заряд того же знака, что и фракционируемые макромолекулы, но не взаимодействуют с ними. Краситель тоже передвигается в электрическом поле, но уже в виде окрашенной зоны. Его скорость должна быть немного выше скорости миграции наиболее подвижных макромолекул. Когда окрашенная зона доходит до конца разделительной камеры, электрофорез прекращают. При разделении структурных мышечных белков для визуализации фронта ЭФ используют бромфеноловый синий (рис. 1.6).
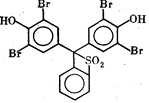
Рис. 1.6. Бромфеноловый синий
Разделившиеся зоны биополимеров во избежание их диффузии немедленно фиксируют. Для этого гель извлекают из стеклянной формы и выдерживают в смеси кислоты со спиртом так, что белки или нуклеиновые кислоты выпадают в осадок в том самом месте, где закончилась их миграция в ходе электрофореза. Одновременно с фиксацией проводят окрашивание зон путем выдерживания геля в растворе красителя, прочно связывающегося с белком или нуклеиновой кислотой. Излишек красителя удаляют. Для окрашивания белков используют краситель Кумасси-синий (рис. 1.7).
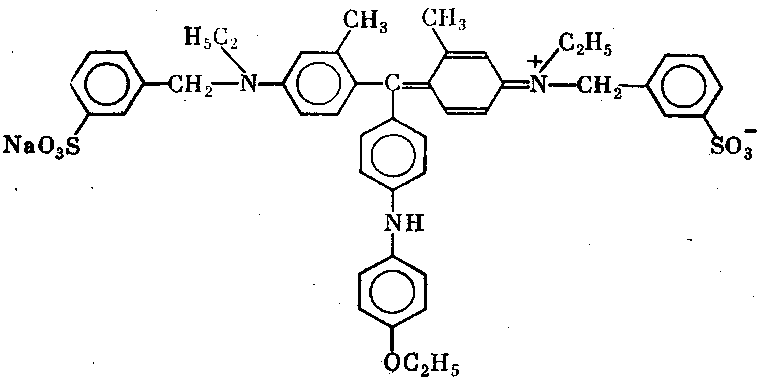
Рис. 1.2. Кумасси G-250.
1.3. Применение
Метод электрофореза, предложенный еще в начале ХХ века, сейчас широко используют в биологии и медицине для разделения белков в исследовательских и клинических целях. С помощью электрофореза можно разделить на отдельные компоненты белковую смесь, что позволяет установить молекулярную массу белка или его субъединиц, подтвердить чистоту выделенного белка. Таким образом, электрофоретический метод в биохимии – это способ пространственного разделения молекул в плотном пористом геле, имеющих разный заряд и размеры. Результатом проведения электрофореза является электрофореграмма – изображение, полученное после разделения сложной смеси с помощью электрофореза и специфического проявления (окрашивания).
Электрофореграммы белков различных биологических жидкостей человека (сыворотка крови, моча, спинномозговая жидкость и др.) позволяют врачам и медицинским биохимикам получить значительную диагностическую информацию. Результаты электрофоретического разделения ферментов (зимограммы) позволяют изучать изменения активности и изоферментного спектра таких белков под действием внешних и внутренних факторов, как у человека, так и у других организмов.
1.4. Преимущества и недостатки
В настоящее время электрофорез в ПААГ является наиболее распространенным способом электрофоретического разделения белков. Популярность метода, во многом, обусловлена его высокой разрешающей способностью и экспериментальной гибкостью. Он позволяет варьировать в широких пределах пористость геля, добиваясь тем самым оптимальных условий при разделении каждого конкретного образца. Путём изменения состава геля и буферных систем можно осуществлять разделение макромолекул по массе, по заряду или по соотношению между их массой и зарядом.
ПААГ прозрачен, химически стабилен, инертен, упругий, прочный, устойчив к изменениям рН и температуры (гели можно кипятить, для более быстрого и четкого окрашивания белков), нерастворим в большинстве растворителей, в нем практически отсутствуют адсорбция и электроосмос.
К недостаткам метода можно отнести использование для создания геля акриламида, который является токсичным веществом и в больших количествах поражает нервную систему, печень и почки.
1.5. Характеристика разделяемых белков
Миозин, сократительный белок поперечнополосатых и гладких мышц, – биологический мотор, преобразующий химическую энергию гидролиза АТФ в механическую работу. Молекула миозина представляет собой гетерогексамер с молекулярной массой 450–500 кДа и длиной около 17 нм, состоящий из двух тяжёлых цепей (200–240 кДа), двух регуляторных (17–21 кДа) и двух существенных (16–22 кДа) лёгких цепей. Эти шесть полипептидных цепей удерживаются между собой за счёт нековалентных связей и образуют комплекс, называемый мономерным миозином.
Актин – глобулярный белок массой 42 кДа – в результате полимеризации образует нить, длина которой может достигать нескольких микрометров. Мономеры актина (их часто называют G-актином) могут полимеризоваться, образуя фибриллярный F-актин. Полимерная нить актина имеет спиральную структуру.
Тропомиозин (Tm) состоит из двух скрученных между собой α-спиралей и имеет вид длинной слабо изогнутой спирали, примерно комплементарной спирали актина; молекулярная масса тропомиозина 30 кД. В скелетных мышцах человека присутствует в виде гомодимеров α- и β-изоформ, а для гладкой мышцы характерно наличие αβ-гетеродимеров.
Тропонин – глобулярный белок с молекулярной массой 80 кД. В его состав входят три субъединицы (Тn-I, Тn-С, Тn-Т). Тn-С (кальций-связывающий) обладает значительным сродством к ионам кальция (молекулярная масса находится в диапазоне от 17 до 20 кД), Тn-I («ингибирующий») может присоединяться к актину (молекулярная масса составляет 33 кД), фиксируя весь тропонин-тропомиозиновый комплекс на его поверхности, и тем самым ингибируя связывание миозиновых головок с актином и их АТФазную активность, либо сам присоединяется к Тn-С, ослабляя связь регуляторных белков с актином. Тn-Т (тропомиозин-связывающий) обеспечивает связь двух других субъединиц между собой и с тропомиозином (молекулярная масса находится в диапазоне от 37 до 40 кД). Каждый тропониновый комплекс связан с одной молекулой тропомиозина.
2. Практическая часть
Электрофорез проводится при температуре 4 ̊С на оборудовании фирмы Bio-Rad.
В качестве объектов исследования использованы мышечные белки, выделенные из соответствующих тканей методом экстракции: миозины предсердий и желудочков свиньи или крысы, скелетной мышцы кролика; актин и тропонин из скелетной мышцы кролика; тропомиозин человека получен в результате экспрессии в E. coli.
2.1. Приготовление рабочих растворов
Приготовить рабочие растворы согласно табл. 2.1.
Таблица 2.1
Рабочие растворы
| Раствор | Концентрация | Масса, г | Деионизированная вода, мл |
| Tris-HCl (pH 8,8) | 1,5 М | 9 | 50 |
| Tris-HCl (pH 6,8) | 1,5 М | 9 | 50 |
| ДСН | 10 % | 1,5 | 15 |
| ПСА | 10 % | 0,1 | 1 |
Для приготовления Tris-HCl в стакан внести 9 г Tris (Base), растворить его в 50 мл деионизированной воды и довести до нужного значения pH с помощью раствора разбавленной соляной кислоты.
ДСН – мелкодисперсный порошок, поэтому при взвешивании реагента и приготовлении раствора необходимо соблюдать осторожность. Также ДСН является ПАВ, т.е. сильно пенится, следовательно, нельзя включать мешалку на большую скорость.
2.2. Ход работы
1. Для работы необходимо набрать 1,5 л дистиллированной воды и поставить ее в холодильник.
2. Подготовить камеры: стекла скрепить при помощи держателя и установить на заливочный столик, полученные камеры проверить на герметичность, заполнив деионизированной водой.
3. Приготовить разделяющий гель по табл. 2.2: TEMED и APS добавить в последний момент.
Таблица 2.2.
Состав разделяющего геля 10 %
| Компонент | мл | мкл |
| Бис-акриламид (30%) | 4,7 | 4700 |
| 1,5 М Tris-HCl (pH 8,8) | 3,5 | 3500 |
| ДСН | 0,14 | 140 |
| Деионизированная вода | 5,0 | 5000 |
| TEMED | 0,007 | 7 |
| ПСА | 0,0696 | 69,6 |
| Объем | 14 | 14000 |
4. Слить дистиллированную воду из камер.
5. Приготовленный гель аккуратно залить в пространство между стеклами, сразу после этого поверх геля наслоить изопропанол (120 мкл) для выравнивания поверхности геля. Гели поставить в теплое место для полимеризации на 60 мин.
6. Приготовить раствор трис-глицинового (стокового) буфера (табл. 2.3). для приготовления буфера используется охлажденная дистиллированная вода из холодильника, приготовленная вначале занятия. Раствор стокового буфера закрыть фольгой и убрать в холодильник.
Таблица 2.3.
Состав трис-глицинового буферного раствора
| Компонент | на 1000 мл |
| Tris | 3 г |
| Глицин | 14,4 г |
| ДСН | 10 мл |
| H2O | 1 л |
7. Подготовить гребенки, протерев их спиртом.
8. Разделяющий гель промыть от изопропанола деионизированной водой.
9. Приготовить концентрирующий гель по табл. 2.4.
Таблица 2.4.
Состав концентрирующего геля
| Компонент | % | мл | мкл |
| Бис-акриламид (30%) | 4,7 | 0,588 | 588 |
| 1,5 М Tris-HCl (pH 6,8) | 0,416 | 416 | |
| SDS | 0,05 | 50 | |
| 0,05% TEMED | 0,005 | 5 | |
| 0,1% APS | 0,05 | 50 | |
| H2O | 3,891 | 3891 | |
| Объем | 5 | 5000 |
10. Полученный гель (1,5 мл) залить поверх разделяющего геля, аккуратно вставить протертые спиртом гребенки. Поставить в теплое место на 30 мин.
11. Приготовить пробы белка в эппендорфах объемом 0,6 мл, для этого исследуемые белки необходимо развести до концентрации 1 мг/мл буферным раствором (sample buffer), содержащим краситель бромфеноловый синий, добавляемый для отслеживания процесса ЭФ (табл. 2.5). Содержимое эппендорфов суспендировать,. поставить в термостат для денатурации белков при температуре 80–90 ̊С на 2–3 мин.
Таблица 2.5.
Состав sample buffer для разведения проб
| Компонент | Объем, мкл |
| 1,5 М Tris-HCl (pH 6,8) | 208,3 |
| 1 М ДТТ | 500 |
| (1-5)% 2-меркаплоэтанол | 50 |
| 0,04 % бромфеноловый синий | 25 |
| 10% ДСН | 1000 |
| Глицерол | 500 |
| Деионизированная вода | До 5 мл |
12. Собрать установку, для этого стекла с готовым гелем закрепить в специальном держателе таким образом, чтобы получилась герметичная емкость для стокового буферного раствора. Из стекол аккуратно убрать гребенки (получатся прямоугольные лунки вместимостью по 15–20 мкл). В образовавшуюся камеру залить стоковый буферный раствор (200 мл), с помощью дозатора промыть лунки буфером.
13. В тетради составить схему заполнения лунок (табл. 2.6), согласно которой внести пробы, указать схему в отчете.
Таблица 2.6.
Схема заполнения лунок в концентрирующем геле
1 гель:
| № лунки | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| мкл | ||||||||||
| проба |
2 гель:
| № лунки | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| мкл | ||||||||||
| проба |
14. Оставшийся раствор буфера залить в емкость аппарата.
15. Аппарат закрыть крышкой и установить на лед. Выставить режим:
a) А = 20 мА, t = 20 минут
b) А = 40 мА, t = 40 минут
В зависимости от условий время этапов ЭФ может варьироваться, поэтому необходимо визуально оценивать скорость прохождения красителя в геле (когда пробы сконцентрировались и дошли до границы между концентрирующим и разделяющим гелем, можно переходить ко второму этапу ЭФ).
16. По окончании электрофореза, когда краситель дошел до нижнего края стекла, выключить установку и аккуратно достать гели. Перенести его в емкость для окрашивания, в ней предварительно гель промыть от SDS водой (можно водопроводной), воду слить, гель залить окрашивающим раствором Кумасси-синий (табл. 2.7).
Таблица 2.7.
Приготовление окрашивающего раствора Кумасси-синий
| Компонент | Количество |
| Бриллиантовый синий Р | 1 г |
| Этиловый спирт | 50 мл |
| Ледяная уксусная кислота | 20 мл |
| Деионизированная вода | 130 мл |
Гель выдерживается в красителе в течение суток.
17. После окрашивания гель заливается отмывочным раствором (табл. 2.7).
Таблица2.7.
Состав отмывочного раствора
| Компонент | Содержание, % | Кол-во, мл |
| Ледяная уксусная кислота | 10 | 50 |
| Этиловый спирт | 20 | 100 |
| H2O | До 500 (≈ 350) |
Отмывку проводить в несколько подходов. Раствор для отмывки сливать сразу, как только его цвет станет таким же, как цвет геля. Гель отмывается пока не станет прозрачным, а пробы четкими.
2.3. Интерпретация результатов
После покраски и отмывки на электрофореграмме четко проявляются окрашенные белковые полосы, расположенные на разном расстоянии от линии раздела концентрирующего и разделяющего гелей. Расстояние зависит от молекулярной массы субъединиц белков, чем меньше масса, тем ближе полоса к нижнему краю разделяющего геля.
Для определения наличия фракций белка во время электрофореза используют маркеры, которые представляют собой готовые смеси белков с известными молекулярными массами (рис. 2.1–А).
В качестве примера на рис. 2.1–Б представлены электрофореграммы различных белков:
1. α-Тропомиозин (α-Tm) – чистая проба белка, поэтому одна полоса в районе 30 кДа.
2. F-актин – полоса соответствует полосе маркера с массой 40 кДа.
3. Тонкий филамент (Thin filament) – комплекс белков, содержащий актин, тропомиозин, тропониновый комплекс. Четко различается полоса, соответствующая F-актину (40 кДа), чуть ниже лежит полоса – тропонин Т (TnT), полоса тропомиозина (30 кДа), ниже лежать полосы тропонина I (TnI) – 25 кДа, тропонина С (TnC) – 18 кДа.
4. Человеческий миозин правого предсердия (human RA): существенные легкие цепи (ELC) – 25 кДа, регуляторные легкие цепи (RLC) – 20 кДа.
5. Человеческий миозин левого желудочка (human LV): существенные легкие цепи (ELC) – 18 кДа, регуляторные легкие цепи (RLC) – 15 кДа. Образец загрязнен актином – 40 кДа.
6. Свиной миозин левого желудочка (Pig LV): существенные легкие цепи (ELC) – 20 кДа, регуляторные легкие цепи (RLC) – 15 кДа. Образец загрязнен актином – 40 кДа.
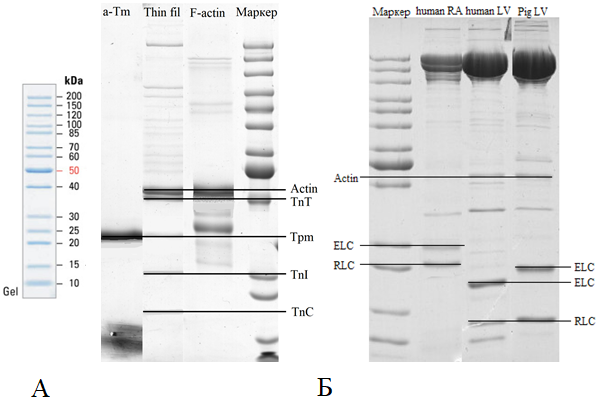
Рис. 2.1. Электрофореграмма маркерной смеси ThermoFisher Unstained Protein Ladder
По рис. 2.1 заметно, что существенные цепи лежат выше регуляторных, при этом легкие цепи предсердия лежат выше, т.е. они тяжелее, чем легкие цепи желудочка. Молекулярные массы свиных и человеческих регуляторных цепей левого желудочка совпадают, существенные легкие цепи свиного левого желудочка тяжелее человеческих, поэтому лежат выше на электрофореграмме.
2.4. Подготовка отчета
Отчет по лабораторной работе «Электрофорез по Лэммли» должен содержать краткую теоретическую часть с описанием назначения используемых реагентов, структурную формулу фрагмента ПААГ, подробное описание выполненных работ (с указанием использованных лабораторных инструментов и реактивов), а также выводы по проделанной работе (с приложением изображений электрофореграмм и интерпретацией результатов).
c) Контроль знаний
Для получения допуска к проведению лабораторной работы необходимо решить тест, предназначенный для объективного оценивания уровня подготовки студентов по теме «Электрофорез в полиакриламидном геле».
Тест включает в себя 10 вопросов, из которых 2 – открытого типа (за правильный ответ 2 балла, за неправильный – 0), 8 – с выбором правильного ответа (правильный ответ – 1 балл, неправильный – 0). Всего за тест можно получить 12 баллов. К выполнению лабораторной работы допускаются студенты, набравшие не менее 8 баллов.
3.1. Контрольные вопросы