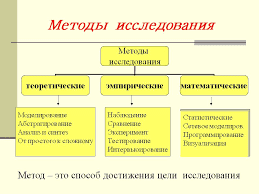
2020-10-10
2020-10-10 121
121Антителопродукции
Составляют 50 - 70% общего количества ПИД. Эта группа ПИД включает девять различных форм (табл. 12).
Таблица 12
ПИД с преимущественным нарушением антителопродуции
| Форма | Патогене-тический механизм | Уровень Ig сыворотки | Число В-клеток | Тип наследования | Сопутствующие признаки |
| 1. Сцепленная с х-хромосомой агаммаглобулинемия (СХАГГ) | Мутация в гене Btk | Снижены все изотипы | Резкое уменьшение | ХС | |
| 2. Селективные дефициты Ig а) субклассов IgG б) IgА | Дефекты дифференцировки изотипов Дефект терминальной дифференцировки в IgA+ В-клетки | Снижен один изотип IgG или более Снижены IgА1 и IgА2 | В норме или незрелые В норме или уменьшены sIgA+ В-клетки | Не известен Вариабелен | Аутоим-мунные и аллергические проявления |
| 3. Общая вариабельная иммунная недостаточность (ОВИН) | Вариабельный, не определен | Снижено несколько изотипов | В норме или уменьшено | Вариабелен |
Окончание табл. 12
| Форма | Патогене-тический механизм | Уровень Ig сыворотки | Число В-клеток | Тип наследования | Сопутствующие признаки |
| 4. Иммунодефицит с повышенным уровнем IgМ | Не известен | IgМ, IgD повышены или в норме, остальные низкие | Есть IgМ, IgD-несущие клетки, отсутствуют другие | АР | Нейтропения, тромбоцитопения, гемолитическая анемия, поражение ЖКТ, печени |
| 5. Транзиторная младенческая гипогаммаглобулинемия | Замедленное созревание хелперной функции | Снижены IgG, IgА | В норме | Не известен | Часто наблюдается в семьях с другими иммунодефицитными состояниями |
Особого внимания из рассматриваемой группы ПИД заслуживают Х-сцепленная агаммаглобулинемия, описанная самой первой среди других ПИД O.Bruton (1952), и наиболее часто встречающиеся формы – ОВИН, селективный дефицит IgA.
Х-сцепленная агаммаглобулинемия, или болезнь Брутона (1:50 000), наблюдается у мальчиков, проявляясь на 5 - 9-м мес. жизни, когда происходит истощение трансплацентарно полученных материнских Ig. Заболевание проявляется рецидивирующими пиогенными инфекциями (пневмонии, синуситы, мезотимпаниты, менингиты). Важный диагностический симптом – лимфатические узлы, селезенка не реагируют увеличением на воспалительный процесс. При иммунолабораторном исследовании выявляют: 1) снижение или отсутствие γ-глобулинов в сыворотке крови; 2) снижение уровня сывороточных IgG (менее 2 г/л) при отсутствии или резком снижении уровней IgМ и IgА; 3) отсутствие или резкое уменьшение числа В-лимфо-цитов (CD19+ или CD20+) в циркуляции менее 2 %; 4) отсутствие или гипоплазию миндалин; 5) малые размеры лимфоузлов; 6) сохранную функцию Т-лимфоцитов.
ОВИН (1:10 000 – 1:50 000)–гетерогенная группа болезней с дефектом антителообразования и различным типом наследования (RosenF.S. и соавт., 1997). Термин «вариабельный» означает манифестацию заболевания в разном возрасте (детском, подростковом, взрослом) с индивидуальными вариациями типа и степени выраженности иммунодефицита. По клинической картине ОВИН напоминает болезнь Брутона, основное отличие – в сроке манифестации заболевания: средний возраст клинического проявления ОВИН – 25 лет, а постановки диагноза – 28 лет (BallowM., 2003). Показатель смертности через 25 лет после диагностирования заболевания – 24 %, основные причины смерти – лимфома (18 %), хронические заболевания легких (11 %). Выживаемость пациентов зависит от степени снижения уровня IgG и недостаточности клеточного звена иммунного ответа: чем более они выражены, тем раньше погибают больные ОВИН. Эта форма ПИД в равной степени поражает мужчин и женщин. Как и все гуморальные иммунодефициты, ОВИН клинически проявляется рецидивирующими и хроническими пневмониями, синуситами, отитами, часто формируются бронхоэктазы, в половине случаев поражается желудочно-кишечный тракт с симптомами мальабсорбции, снижением массы тела, диареей, гипоальбуминемией, дефицитом витаминов. Характерны хронические воспалительные процессы в кишечнике (энтеровирусные инфекции) с развитием нодулярной лимфоидной гиперплазии. Порядка одной трети больных имеют спленомегалию и/или диффузную лимфаденопатию. В 22 % случаев развиваются аутоиммунные проявления (пернициозная или гемолитическая анемия, тромбоцитопения, нейтропения, ревматоидный артрит, нарушение функции щитовидной железы). При иммунолабораторном исследовании выявляют: 1) нормальное или несколько уменьшенное число циркулирующих В-лимфоцитов; 2) снижение уровней сывороточных IgG и IgA, в меньшей степени уровня IgM; снижение суммарной концентрации IgG + IgA+IgM менее 3 г/л; 3) общее число Т-клеток в норме или несколько уменьшено за счет сокращения численности Т-хелперной субпопуляции; 4) снижен иммунорегуляторный индекс CD4+/CD8+.
Селективный дефицитIgA (1:700 у европеоидов; 1:18 500 у японцев) характеризуется снижением уровня сывороточного IgA до 0,05 г/л и ниже (довольно часто до 0) при нормальном содержании других классов Ig. Если концентрация IgA выше 0,05 г/л, но ниже 0,2 г/л, то следует выставлять диагноз «парциальная (частичная) недостаточность IgA». В большинстве случаев дефицит IgA протекает бессимптомно,.однако у части индивидов проявляется синопульмональными инфекциями в сочетании с аллергическими проявлениями (атопический дерматит, поллиноз, бронхиальная астма, отек Квинке и др.) и аутоиммунными (склеродермия, ревматоидный артрит, витилиго, тиреоидит). При обследовании 240 практически здоровых жителей сельских районов Чувашии, проведенном в летние месяцы (июнь - июль), мы выявили отсутствие IgA у двух индивидов (мужчины 18 лет и женщины 42 лет). Вторая серия исследований, включавшая иммунологическое тестирование другой группы практически здоровых людей и осуществленная в зимний период (январь - февраль), не обнаружила дефицита ни у одного обследованного. Мы объясняем этот факт следующим. В группу практически здоровых отбирались лица, не имевшие хронических заболеваний и не болевшие в предшествовавшем исследованию месяце ОРЗ. Зимний период в нашем регионе является фактором риска развития респираторных инфекций, и, видимо, лица с дефектом выработки IgA, восприимчивые кразличного рода инфекциям, имели эпизоды ОРЗ и не попали в группу здоровых. В связи с этим интересны данные, в которых сообщается о том, что при обследовании 2025 здоровых доноров в Малайзии не обнаружено ни одного случая селективного дефицита IgA. Авторы это связывают с тем, что в условиях широкого распространения инфекции в тропиках отсутствие IgA не может протекать бессимптомно, обязательно проявится клинически какой-либо инфекцией.
Транзиторная гипогаммаглобулинемия у детей («медленный иммунологический старт») характеризуется низкими уровнями Ig. Начало заболевания с 5 - 6 мес., когда ребенок внезапно, без видимых причин начинает болеть рецидивирующими пиогенными инфекциями почек, респираторного тракта. Это связано с тем, что материнские IgG, полученные ребенком трансплацентарно, к этому возрасту катаболизировались, а продукция собственных IgG, обычно начинающаяся с 4-го мес. жизни ребенка, запаздывает. При этой форме иммунодефицита чаще снижены уровни IgG и IgА, в то время как уровень IgМ находится в пределах нормы или даже повышен. В-лимфоциты, лимфоузлы и миндалины не изменены. Такое транзиторное иммунодефицитное состояние встречается у 5 - 8 % грудных детей (обычно у недоношенных детей или детей из семей с иммунодефицитами) и обычно проходит без лечения к 1,5 - 4 годам.