Мышцы характеризуются высоким обменом белков и АК. Белки и АК в мышцах активно синтезируются и распадаются.
Белок скелетных мышц является важным источником АК для всего организма. В условиях голодания и энергодефицита белки мышц разрушаются, а образовавшиеся АК покидают мышцы и активно используются организмом в качестве источника энергии.
У млекопитающих мышцы являются главным местом катаболизма АК с разветвленной цепью. Мышечная ткань окисляет лейцин до СО2 и превращает углеродный скелет аспартата, аспарагина, глутамата, изолейцина и валина в субстраты ЦТК. Способность мышц разрушать АК с разветвленной цепью при голодании и диабете возрастает в 3— 5 раз.
Мышцы также синтезируют и выделяют много аланина и глутамина. В синтезе этих АК используются аминогруппы, которые образуются при распаде АК с разветвленной цепью и затем переносятся на α-КГ и ПВК в ходе реакций трансаминирования. Источником почти всего пирувата, идущего на синтез аланина, является гликолиз (глюкозо-аланиновый цикл).
При интенсивной работе мышцы выделяют аммиак. В мышечной ткани активность глу-ДГ низка, поэтому при интенсивной работе функционирует в основном путь непрямого дезаминирования с участием цикла ИМФ-АМФ.
ФОРМУЛА!
Болезнь МакАрдла (тип V) — аутосомно-рецессивная патология, отсутствует в скелетных мышцах активность гликогенфосфорилазы. Накопление в мышцах гликогена аномальной структуры.
Энергетический обмен
Энергетический обмен в мышцах отличается от всех тканей тем, что в состоянии покоя он очень низкий, а при интенсивной физической нагрузке он значительно возрастает.
Различия энергетического обмена наблюдаются и в самих мышцах. В белых (белых) волокнах преобладает анаэробный гликолиз, субстратом которого является только глюкоза. В красных (медленных) мышцах преобладает аэробное окисление жирных кислот, кетоновых тел и глюкозы.
Миокард в норме в качестве субстратов для синтеза АТФ использует жирные кислоты (65 — 70%), глюкозу (15 — 20%) и молочную кислоту (10 — 15%). Роль аминокислот, кетоновых тел и пирувата в энергообеспечении миокарда сравнительно невелика.
Основным потребителем АТФ в мышечной ткани является процесс мышечного сокращения. Запасы АТФ в скелетной мышце при сокращении быстро истощаются, и их хватает менее чем на секундное сокращение.
Для того, чтобы обеспечить интенсивно работающую мышцу достаточным количеством энергии, в мышце существует несколько источников АТФ.
1. АТФ образуется по классическому пути в реакциях субстратного и окислительного фосфорилирования.
2. АТФ образуется из 2 АДФ при участии миоаденилаткиназы: АДФ + АДФ → АТФ + АМФ;
3. АТФ образуется при работе креатинфосфатного челнока.
Креатинфосфатный челнок
В работе креатинфосфатного челнока участвуют креатинфосфат, креатин и изоформы фермента креатинфосфокиназы (КФК).
Синтез креатина в основном происходит в печени из 3 АК: аргинин, глицин и метионин. Из печени креатин с током крови поступает в мышечную ткань, а также в нервную ткань.
· Образованная в процессе окислительного фосфорилирования АТФ переносится АТФ/АДФ-транслоказой через внутреннюю мембрану митохондрий.
· В межмембранном пространстве митохондрий АТФ с участием и митохондриальной креатинкиназы фосфорилирует креатин в креатинфосфат: АТФ + креатин → АДФ + креатинфосфат
· Затем креатинфосфат направляется к миофибриллам (или к другим местам потребления энергии).
· Под действием креатинкиназы миофибрилл креатинфосфат фосфорилирует АДФ в АТФ: АДФ + креатинфосфат → АТФ + креатин
· Образующийся креатин снова возвращается к митохондриям и цикл повторяется.
Работа креатинфосфатного челнока предотвращает быстрое истощение запасов АТФ в мышце. Это происходит благодаря тому, что:
1. в креатинфосфате создается запас макроэргических связей;
2. креатинфосфат меньше АТФ и по этому он гораздо подвижнее. Он гораздо быстрее доставляет энергию от митохондрий к работающей миофибрилле, чем АТФ (скорость примерно на три порядка выше). При этом, скорость доставки энергии с помощью креатинфосфата заведомо превышает максимльную скорость ее использования.
Существует несколько изоферментов КФК не только в разных органах, но и в одной и той же клетке. В частности, в мышечных клетках идентифицированы четыре изофермента — в митохондриях, миофибриллах, мембранах
Тропонины – глобулярные белки, которые образуют тропониновую систему.
Тропонин I (TпI) ингибирует взаимодействие между F-актином и миозином и также связывается с другими компонентами тропонина.
Тропонин С (ТпС) — кальций-связывающий белок с массой 17000Да, он может связывать 4 Са2+, его строение и свойства аналогичны кальмодулину.
Тропонин Т (ТпТ) как и другие тропонины, связывается с тропомиозином.
α-Актинин – белок, который образует в миофибрилле Z-диск.
Строение миофибриллы
Миофибрилла состоит из одинаковых повторяющихся элементов - саркомеров.
Саркомер - функциональная единица миофибриллы, он имеет длину от 1500 до 2300 нм.
· Саркомер ограничен с двух сторон Z-дисками, образованные α-актинином.
· К Z-дискам присоединены «тонкие» филаменты. Тонкие филаменты гладких мышц образованы F-актином и тропомиозином, а поперечнополосатых - F-актином, тропомиозином и тропонинами Т, I и С. Диаметр тонких филаментов составляет около 6 нм.
· В центре саркомера, между «тонкими» филаментами, располагаются «толстые» филаменты. «Толстые» филаменты имеют диаметр около 16нм, они образованы молекулами миозина. На поверхности «толстого» филамента с промежутками в 14 нм располагаются головки миозина, с помощью которых «толстые» филаменты взаимодействуют с актином «тонких» филаментов. В центре «толстых» филаментов на участке в 150 нм миозиновых головок нет.
Каждый «тонкий» филамент занимает симметричное положение между тремя толстыми филаментами, а каждый «толстый» филамент симметрично окружен шестью «тонкими» филаментами.
Расположение филаментов в поперечнополосатой мышце (по Р. Марри, 1993).
В скелетной мышечной ткани мышечные волокна выстраивается таким образом, что саркомеры миофибрилл располагаются параллельно. При этом на срезах наблюдается правильное чередование светлых и темных участков, благодаря которым скелетные мышцы называют поперечнополосатыми.
· Темный участок – называется диск А (анизотропная зона), он образован «толстыми» нитями миозина. Его размер постоянен.
· Центральная область диска А называется зона Н, она выглядит менее плотной, чем остальная его часть. В зоне Н нет «тонких» нитей актина, в отличие от более темной части, которая образована и «толстыми» и «тонкими» нитями. Размер зоны Н уменьшается при сокращении мышцы.
· Полоса М пересекает центральную область диска А, она образована толстыми нитями, в которых миозин не имеет головок. Полоса М имеет длину 150 нм, в не заходят «тонкие» нити актина.
· Светлый участок называется диск I (изотропная зона), он образован «тонкими» нитями актина. Размер диска I уменьшается при сокращении мышцы. Диск I делит пополам очень плотная и узкая линия Z, которая образована Z-дисками α-актинина.
Механизмы мышечного сокращения
Мышечное сокращение состоит из циклов присоединения и отсоединения глобулярной «головки» миозина от нити F-актина. Биохимический цикл мышечного сокращения состоит из пяти стадий:
При действии, например, ацетилхолина на ацетилхолиновые рецепторы происходит возбуждение сарколеммы.
Потенциал действия сарколеммы, через Т-систему у скелетных мышц или напрямую у миокарда и гладких мышц, достигает кальциевых каналов саркоплазматического ретикулума (рианодиновые рецепторы).
Кальциевые каналы открываются, выпуская Са2+ из саркоплазматического ретикулума в саркоплазму, так что его концентрация в ней возрастает до 10-5 моль/л.
Далее механизм регуляции мышечного сокращения в поперечнополосатых и гладких мышцах отличается.
Актиновая регуляция
Актиновая регуляция характерна для поперечнополосатых мышц - скелетных и сердечной.
Мышечное сокращение скелетных мышц ингибирует тропомиозиновая система на 2 стадии сокращения, так как TпI предотвращает присоединение миозиновой головки к соответствующему связывающему сайту F-актина (TпI или изменяет конформацию F-актина или перемещает тропомиозин в то положение, в котором он блокирует сайты связывания миозиновых головок на F-актине).
Поступающий в саркоплазму Са2+ присоединяется к тропонину ТnС. Комплекс ТnС•Са2+ реагирует с TnI и ТnТ, влияя на их взаимодействие с тропомиозином. Тропомиозин при этом либо отсоединяется, либо изменяет конформацию F-актина таким образом, что появляется возможность присоединения к нему миозиновой головки тяжелой цепи. Начинается сократительный цикл.
Расслабление происходит, когда 1) содержание Са2+ в саркоплазме падает ниже 10-7 моль/л вследствие его поглощения саркоплазматическим ретикулумом; 2) комплекс ТnС•Са2+ утрачивает свой Са2+; 3) тропонин, реагируя с тропомиозином, ингибирует дальнейшее взаимодействие миозиновой головки с F-актином и 4) миозиновые головки в присутствии АТФ отделяются от F-актина, вызывая расслабление.
Так как в сердечной мышце основным источником ионов Са2+ для возбуждения служит внеклеточная жидкость, при отсутствии Са2+ во внеклеточной жидкости сокращения сердечной мышцы прекращаются в течение одной минуты. Скелетная мышца в таких условиях может сокращаться часами.
Исчезновение АТФ из саркоплазмы приводит к следующим последствиям: 1) Са2+-насос саркоплазматического ретикулума перестает поддерживать низкую концентрацию Са2+ в саркоплазме; при этом стимулируется взаимодействие миозиновых головок с F-актином; 2) не происходит зависимого от АТФ отделения миозиновых головок от F-актина, при этом на 5 стадии мышечного сокращения наступает трупное окоченение.
Миозиновая регуляцияМиозиновая регуляция характерна для гладких мышц. У гладких мышц нет тропониновой системы, а легкая цепь (р-цепь) миозина подавляет его АТФ-азную активность и препятствует присоединению миозина к F-актину.
В саркоплазме гладких мышц присутствует киназа легких цепей миозина, зависимая от Са2+. При повышении в саркоплазме Са2+, он присоединяется к кальмодулину. Комплекс кальмодулин-4Са2+ активирует киназу легких цепей миозина.
Активная киназа легких цепей миозина фосфорилирует легкую цепь р, которая при этом перестает ингибировать АТФ-азную активность миозина и препятствовать взаимодействию миозина с F-актином. В результате начинается со
С-реактивный белок (СРБ)
Положительные результаты серологического определения СРБ обычно наблюдаются при бактериальной инфекции, инфаркте миокарда, злокачественных опухолях, лимфогранулематозе, нефрите, а также при отдельных формах коллагенозов: ревматизме, красной волчанке, инфекционном неспецифическом полиартрите.
Креатин
Креатинурия появляется при патологических состояниях мышечной ткани: миопатии или прогрессирующей мышечной дистрофии.
При повреждениях мышц снижено содержание калия в крови, повышено содержание аминокислот в моче.
Основные нарушения обмена веществ различных видов мышечной ткани, причины, последствия, биохимическая диагностика
Миопатии и миодистрофии
Миопатии (греч. mys, myos мышца + pathos страдание, болезнь) - нервно-мышечные заболевания, характеризующиеся прогрессирующим развитием первичного дистрофического или вторичного (денервационного) атрофического процесса в скелетной мускулатуре, сопровождающиеся мышечной слабостью и двигательными нарушениями.
К миопатиям относят как наследственные нервно-мышечные заболевания, так и разнообразные нервно-мышечные синдромы при ряде соматических и неврологических болезней.
Наследственные миопатии, в основе которых лежат первичные обменные нарушения и расстройства микроциркуляции в мышечной ткани, приводящие к дистрофии мышечных волокон с замещением их соединительной и жировой тканью, относят к группе прогрессирующих мышечных дистрофий (см. Дистрофии мышечные прогрессирующие), а наследственные миопатии, обусловленные нарушением иннервации мышц вследствие поражения сегментарных мотонейронов спинного мозга или периферических нервных волокон, - к группе спинальных или невральных амиотрофий.
В основе развития миопатии лежат нарушение обмена в мышечных клетках, изменение синтеза нуклеиновых кислот, значительное преобладание ускоренного распада белков мышц над измененным их синтезом. Мышцы при миопатии истончены, часть волокон замещена жировой тканью; при электронной микроскопии обнаруживают изменение структуры мембран мышечных клеток. Основные признаки миопатии - нарастающая мышечная слабость, симметричная атрофия мышц, снижение сухожильных рефлексов, в поздних стадиях - деформация костей и суставов. Постоянно выражены вегетативнотрофические расстройства.
Ишемическая болезнь сердца
ИБС патологическое состояние, характеризующееся абсолютным или относительным нарушением кровоснабжения миокарда вследствие поражения коронарных артерий сердца.
Причины поражения коронарных артерий сердца обусловлены наследственными (дефекты сосудов) и воспалительными (васкулиты, большие коллагенозы, инфекционные поражения, например сифилис), так и обменными (атеросклероз, 97-98%) заболеваниями.
Также причиной ишемии может быть нарушение нервной регуляции артерий, приводящее к их спазму.
Классификация. Выделяют пять форм ишемической болезни сердца: 1) первичная остановка кровообращения; 2) стенокардия; 3) инфаркт миокарда; 4) сердечная недостаточность; 5) аритмии.
Основное внимание привлечено к инфаркту миокарда - самой тяжелой и распространенной острой форме ишемической болезни сердца.
выход ионов калия из ишемизированных кардиомиоцитов, накопление в них натрия, кальция, а также жидкости. В отдаленных от зоны ишемии участках сердца концентрация указанных и других ионов, а также жидкости тоже меняется, однако степень этих изменений значительно меньшая.
В качестве ведущих причин К+ — Na+ дисбаланса при КН называют дефицит АТФ, повышение проницаемости сарколеммы и торможение активности К+Ма2+-зависимой АТФазы, что создает возможность пассивного выхода К+из клетки и входа в нее Na+ по градиенту концентрации. КН сопровождается также высвобождением больших количеств К+ и Са2+ из митохондрий. Непосредственными факторами, обусловливающими этот процесс, могут быть снижение мембранного потенциала деэнергизированных митохондрий и увеличение проницаемости их мембраны под влиянием ацидоза, продуктов СПОЛ и фосфолипаз, активируемых Са2+. Значительное количество К+ высвобождается и при гликолитиЧеском распаде молекул гликогена (синтез которого идет с захватом ионов калия).
Потеря К+ клетками миокарда при КН сопровождается повышением содержания его в крови. Гиперкалиемия является характерным признаком КН, особенно завершающейся развитием инфаркта миокарда. В эксперименте на собаках уже в первые пять минут ишемии содержание калия в крови, оттекающей как от ишемизированной, так и отдаленной зон, существенно увеличивается.
Нарушение энергообеспечения кардиомиоцитов, повреждение их мембран и ферментов, дисбаланс ионов и жидкости в совокупности обусловливают расстройство механизма регуляции объема клеток миокарда при КН. Последнее является результатом повышения проницаемости клеточных мембран для ионов и органических гидрофильных молекул (белка, углеводов); гиперосмии кардиомиоцитов в результате накопления в них ионов (натрия, кальция) и мелкодисперсных соединений (альбуминов, пирувата, лактата); гипергидратации и набухания клеток; снижение механической прочности биологических мембран.
Расстройство механизмов регуляции функции сердца
Изменение функции сердца в целом, а также характер и степень повреждения отдельных его клеток при КН являются не только результатом прямой альтерации их патогенными факторами ишемии. В значительной мере это обусловлено и расстройством механизмов регуляции сердечной деятельности, которое развивается преимущественно на одном (реже) или нескольких (чаще) уровнях:
на уровне взаимодействия биологически активных веществ (гормонов, нейромедиаторов) с рецепторами. Изменение чувствительности, числа и(или) конформации молекул рецепторов, их липидного окружения в биологических мембранах может существенно модифицировать характер клеточного ответа на регулирующий стимул; на уровне клеточных «посредников» (мессенджеров) регуляторных влияний, в частности циклических нуклеотидов (цАМФ, цГМФ), образующихся в ответ на действие «первых посредников» — нейромедиаторов и гормонов;
на уровне метаболических клеточных реакций, регулируемых циклическими нуклеотидами и другими внутриклеточными «медиаторами».
КН характеризуется фазными изменениями активности механизмов регуляции, в том числе — симпатической и парасимпатической. На начальном этапе ишемии миокарда, как правило (хотя и не всегда), наблюдается значительная активация
Неизменяемые
Изменяемые
• Наследственность. Риск заболеваемости увеличивается в 2 - 5 раз.
• Пол. Мужской. Но после менопаузы женщины болеют так же часто, как и мужчины, в связи с уменьшением количества женского гормона эстрогена.
• Возраст. В процессе старения организма и накопления жировых отложений на стенках сосудов риск заболеваемости увеличивается.
• Повышение артериального давления - увеличивается возможность развития болезни в 5 раз.
• Повышение уровня холестерина - увеличение холестерина на 1% увеличивает риск развития инфаркта миокарда на 2%.
• Сахарный диабет - удваивает риск развития ишемической болезни сердца.
• Курение - риск внезапной смерти увеличивается в 3 раза.
• Недостаточная физическая нагрузка
• Стрессы
 2015-04-30
2015-04-30 521
521



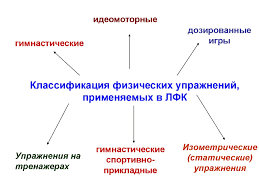

 8537
8537 8173
8173