 2015-05-26
2015-05-26 5695
5695ГЛАВА 5.
5.1. Специфика растворов электролитов.
Теория электролитической диссоциации Аррениуса – Оствальда
5.2. Метод активностей. Элементы теории Дебая – Хюккеля
5.3. Электролитическая диссоциация воды. Водородный показатель
5.4. Теории кислот и оснований
5.5. Произведение растворимости
5.6. Гидролиз солей
Специфика растворов электролитов.
Теория электролитической диссоциации Аррениуса – Оствальда
Электролиты – это химические соединения, которые в растворе или в расплаве самопроизвольно частично или полностью распадаются (диссоциируют) на ионы (от др. греч. ίον – идущий) – заряженные частицы, способные к самостоятельному существованию. Отрицательно заряженные ионы, движущиеся в электрическом поле к положительному электроду (аноду), называют анионами (от др. греч. άνοδος – движение вверх и ίον). Положительно заряженные ионы, движущиеся к отрицательному электроду (катоду), называют катионами (от греч. kάqodoς – ход вниз, возвращение и ίον). Термины «ион», «анион», «катион» ввёл в 1834 г. М. Фарадей. Первоначально предполагали, что ионы образуются в растворе или в расплаве под действием электрического тока (термин «электролиты» возник от др. греч. ήλεκτρον – янтарь, порождающий электричество и λύqος – разлагаемый). Однако впоследствии выяснилось, что в растворах или расплавах, проводящих электрический ток, ионы содержатся независимо от пропускания тока. (Заметим, что в технике электролитами принято называть растворы электролитов.)
Растворы электролитов относятся к проводникам электрического тока второго рода (т. е. ионным), передача электричества осуществляется в них движением как положительных, так и отрицательных ионов (рис. 5.1).
Электрическая проводимость (электропроводность) растворов электролитов, т. е. способность их проводить электрический ток, зависит от природы электролита и растворителя, концентрации, температуры и некоторых других факторов. В электрохимических расчётах часто используют удельную электропроводность.
Удельная электропроводность раствора электролита k – это электропроводность объёма раствора, заключённого между двумя параллельными электродами, имеющими площадь по 1 м2 и расположенными на расстоянии 1 м друг от друга. Удельная электропроводность является величиной, обратной удельному сопротивлению:
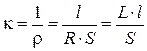 , , | (5.1) |
где r – удельное сопротивление, Ом×м;
R – сопротивление, Ом;
L – электропроводность, 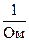 или См (сименс);
или См (сименс);
S – площадь поперечного сечения проводника, м2;
l – длина проводника, м.
k измеряется в 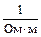 или в
или в 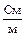 (сименс, делённый на метр).
(сименс, делённый на метр).
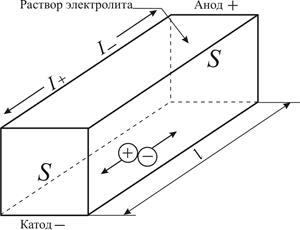 | Рис. 5.1. Схема передачи электричества в слое раствора электролита толщиной l, заключённом между двумя электродами площадью S. Электрический ток в растворе электролита имеет две составляющие: I + (движение катионов к катоду), I – (движение анионов к аноду) |
Итак, растворы электролитов характеризуются электролитической диссоциацией растворённого вещества с образованием ионов. Классическая теория электролитической диссоциации была разработана в 1883 – 87 гг. С.А. Аррениусом Ф.В. Оствальдом, она является первой количественной теорией растворов электролитов.
По современным представлениям суть теории электролитической диссоциации заключается в следующем. Молекулы электролита и растворителя (обычно в качестве такового рассматривают воду) являются полярными. В полярных молекулах электрические центры тяжести положительных зарядов ядер и отрицательных зарядов электронов не совпадают. В молекуле возникает постоянный электрический диполь – электронейтральная система с двумя одинаковыми по величине положительными и отрицательными зарядами q, находящимися на определённом расстоянии друг от друга l (длина диполя). Постоянный электрический диполь обладает постоянным (перманентным) дипольным моментом m = ql, который является мерой полярности связи в молекуле. Дипольный момент измеряют в дебаях D (1 D = 3,336×10–30 Кл×м), эта единица названа в честь одного из основоположников учения о диполях в полярных молекулах П. Дебая. Например, постоянный дипольный момент некоторых полярных молекул m, D: HNO3 (1,94); HF (1,71); H2O (1,65); CH3COOH (1,56); HCl (0,97); HI (0,34) [Карапетьянц]. Дипольный момент является векторной величиной 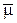 , принято считать, что он направлен от центра тяжести положительного к центру тяжести отрицательного заряда диполя:
, принято считать, что он направлен от центра тяжести положительного к центру тяжести отрицательного заряда диполя:
 .
.
Электролитическая диссоциация – это распад полярных молекул электролитов на ионы под действием полярных молекул растворителя. В кристаллах с ионной связью ионы располагаются в узлах кристаллической решётки. Например, кристаллы NaCl состоят из ионов Na+ и Cl–. При внесении кристалла NaCl в воду ионы Na+ и Cl– переходят в раствор в результате взаимодействия кристалла NaCl с полярными молекулами воды, приводящего к ослаблению электростатического взаимодействия, удерживающего ионы в кристалле и сольватации ионов (от лат. solvo – растворяю) (рис. 5.2.а). При растворении веществ с ковалентной полярной связью в полярных растворителях, имеющих высокую диэлектрическую проницаемость, происходит ослабление и разрыв связей между атомами в молекулах растворяемого вещества, что приводит к образованию ионов (рис. 5.2.б). Например, при растворении в воде серной кислоты под действием молекул воды происходит диссоциация полярных молекул H2SO4 на ионы и сольватация их в растворе.
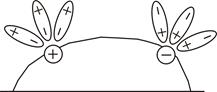 | 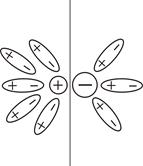 | |
| (а) Отрыв ионов полярными молекулами растворителя от ионного кристалла при его растворении | (б) Диссоциация полярных молекул электролита под действием полярных молекул растворителя |
Рис. 5.2.
Ионы в растворе сольватированы (если растворитель вода – гидратированы, от греч. udwr – вода), т. е. окружены сольватной (гидратной) оболочкой из ориентированных диполей растворителя, которые прочно удерживаются вблизи иона и участвуют в тепловом движении вместе с ионом. Процесс сольватации полярных молекул в общем случае состоит из двух стадий (рис. 5.3). На стадии (а) происходит химическое взаимодействие между молекулами растворяющегося вещества KA и (n + m) молекулами растворителя S с образованием сольватированной молекулы (сольвата) KA (n + m) S:
| KA + (n + m) S «KA (n + m) S. | (5.2) |
На стадии (б) сольват KA (n + m) S диссоциирует на сольватированные ионы Kz + nS и Az – mS:
| KA (n + m) S «Kz+nS + Az–mS. | (5.3) |
Если процесс сольватации останавливается на стадии (а), то растворённое вещество является неэлектролитом (например: спирты и сахара, растворённые в воде). Если процесс сольватации протекает до стадии (б), то растворённое вещество является электролитом (например: неорганические кислоты, основания, соли, растворённые в воде; некоторые соли в жидком аммиаке). Таким образом, для электролитов уравнение сольватации – диссоциации можно записать без промежуточной стадии:
| KA + (n + m) S «Kz+nS + Az–mS. | (5.4) |
Например, процесс диссоциации соляной кислоты HCl в воде (протекает практически необратимо) записывают следующим образом:
HCl + (n + m)H2O ® H+nH2O + Cl–mH2O.
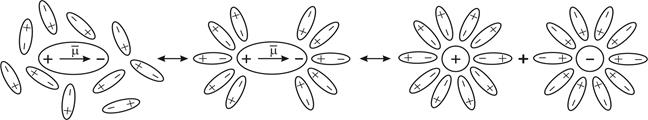 | |
| стадия (а) | стадия (б) |
Рис. 5.3. Схема взаимодействия полярной молекулы растворяемого вещества с полярными молекулами растворителя (сольватации)
Коэффициенты n и m в формулах сольватов меняются с изменением концентрации, температуры и других параметров раствора. Кроме того, между молекулами растворителя в сольватной оболочке и в объёме раствора существует динамическое равновесие. Например, средние времена жизни молекул воды в гидратной оболочке ряда ионов таковы, с: Br– (10–11); Na+ (10–9); Cu2+ (10–7); Fe3+ (10–5); Al3+ (7); Cr3+ (1,5×107 c или 42 ч.) [Чанг]. Поэтому, обычно в уравнениях диссоциации опускают молекулы растворителя (n + m)S. Тогда, уравнение (5.4) будет выглядеть:
| KA «Kz+ + Az–. | (5.5) |
В частности, процесс диссоциации соляной кислоты HCl записывают следующим образом:
HCl ® H+ + Cl–.
Число ионов n, образовавшихся в результате диссоциации одной молекулы электролита, а также величина и знак заряда z этих ионов, зависят от природы электролита. Различают следующие типы электролитов:
1. Бинарные или симметричные электролиты, распадающиеся на 2 иона. Если при этом образуются однозарядные ионы, электролит относят к типу 1,1-зарядных электролитов (NaCl, HCl и др.), если двухзарядные – к типу 2,2-зарядных (MgSO4, ZnSO4 и др.).
2. Тернарные электролиты, распадающиеся на 3 иона. К ним относят 1,2-зарядные (Na2SO4 и др.) и 2,1-зарядные (CaCl2 и др.) электролиты.
3. Квартернарные электролиты, распадающиеся на 4 иона. К ним относят 1,3-зарядные (K3PO4 и др.) и 3,1-зарядные (Al(NO3)3 и др.).
Тернарные и квартернарные электролиты являются несимметричными.
Электролиты при растворении распадаются на ионы не полностью. Только определённая часть растворённых молекул присутствует в виде ионов. Доля молекул, распавшихся в состоянии равновесия на ионы, отвечает степени электролитической диссоциации a, которая равна отношению числа молекул n, распавшихся на ионы, к общему числу растворённых молекул N (ионизированных n и неионизированных n 0):
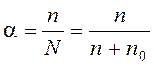 . . | (5.6) |
a может быть выражена в долях единицы или в процентах %.
Степень электролитической диссоциации зависит от природы растворителя, концентрации электролита, температуры и присутствия других электролитов в растворе. Чем выше диэлектрическая проницаемость растворителя, тем больше степень диссоциации электролита (приближённое правило Каблукова – Нернста – Томсона, предложено в 1893г. В.Г. Нернстом и У. Томсоном (Кельвином) на основании данных, полученных ранее И.А. Каблуковым).
a рассматривается теорией электролитической диссоциации как одна из основных количественных характеристик растворов электролитов. По степени диссоциации электролиты разделяют на сильные, средней силы и слабые. К сильным электролитам относят те, у которых a > 30 %. К ним относятся большинство солей (хлорид калия KCl, сульфат аммония (NH4)2SO4, ацетат натрия CH3COONa и др.), многие неорганические кислоты (азотная HNO3, серная H2SO4, хлорная HСlO4, соляная HCl, бромистый водород HBr, йодистый водород HI, и др.) и гидроксиды щелочных металлов (натрия NaOH, калия KOH и др.). Электролиты средней силы имеют степень диссоциации 3 % £ a £ 30 %. К ним относятся некоторые органические и неорганические кислоты (щавелевая (COOH)2, муравьиная HCOOH, сернистая H2SO3, ортофосфорная H3PO4 и др.). Слабые электролиты имеют a < 3 %. К ним принадлежат некоторые кислоты (сероводородная H2S, синильная HCN, метакремниевая H2SiO3, угольная H2CO3 и др.), гидроксиды многих d-металлов (меди Cu(OH)2, хрома Cr(OH)3 и др.), гидроксид аммония NH4OH, немногие соли неорганических кислот (хлорид ртути HgCl2, хлорид кадмия CdCl2, роданид железа (III) Fe(SCN)3 и др.), некоторые соли большинство органических кислот (уксусной CH3COOH, бензойной C6H5COOH и др.) и оснований (пиридина С5H5N и др.), фенолы (гидрохинон C6H4(OH)2 и др.), амины (анилин С6H5NH2 и др.). Все указанные значения a относятся к децимолярному (СМ = 0,1 М) водному раствору [Карапетьянц]. Н.А. Измайлов доказал, что сильные и слабые электролиты являются двумя различными состояниями химических соединений в зависимости от природы растворителя. В одном растворителе данный электролит может быть сильным, в другом – слабым.
В растворах электролитов относительное понижение давления насыщенного пара растворителя 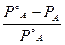 , повышение температуры кипения DТ к, понижение температуры замерзания DТ з, осмотическое давление p значительно больше, чем в растворах неэлектролитов той же концентрации. В связи с этим Я. Вант-Гофф в 1887 г. предложил ввести эмпирический коэффициент i, на который следует умножать величину концентрации раствора электролита, чтобы получить соответствие между экспериментальными величинами , DТ к, DТ з, p и рассчитанными по формулам (4.9), (4.12), (4.13), (4.15) соответственно. Поэтому для растворов электролита указанные формулы принимают вид:
, повышение температуры кипения DТ к, понижение температуры замерзания DТ з, осмотическое давление p значительно больше, чем в растворах неэлектролитов той же концентрации. В связи с этим Я. Вант-Гофф в 1887 г. предложил ввести эмпирический коэффициент i, на который следует умножать величину концентрации раствора электролита, чтобы получить соответствие между экспериментальными величинами , DТ к, DТ з, p и рассчитанными по формулам (4.9), (4.12), (4.13), (4.15) соответственно. Поэтому для растворов электролита указанные формулы принимают вид:
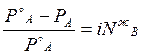 , , | (5.7) | |
| DТ к = iЕСm, | (5.8) | |
| DТ з = iКСm, | (5.9) | |
| p = iСMR T, | (5.10) |
где i – изотонический коэффициент (коэффициент Вант-Гоффа) (i > 1).
Вспомним, что изотоническими называют растворы с одинаковым осмотическим давлением (см. §4.3). Аррениус установил физический смысл i, согласно которому данный коэффициент показывает, во сколько раз увеличилось число частиц в растворе электролита в результате диссоциации.
У слабых электролитов изотонический коэффициент i связан со степенью диссоциации a. Допустим, раствор, содержащий 1 моль (т. е. N A молекул) электролита, имеет степень диссоциации a. Пусть из одной молекулы электролита при диссоциации образуется n ионов, тогда общая концентрация ионов равна n N Aa, концентрация недиссоциированных молекул N A(1 – a). Связь i и a будет выражаться формулой:
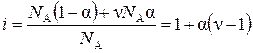 или или 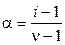 . . | (5.11) |
Из уравнения (5.11) следует, что при a = 0 (раствор неэлектролита) i = 1, а при a = 1 (раствор полностью диссоциированного электролита) i = n. Целочисленные значения i практически не наблюдаются, т. к. уравнение (5.11) не учитывает сил межъионного взаимодействия и др. Однако при a = 0 коэффициент i будет близок к 1, а при a = 1 близок к числу ионов, на которые распадается молекула электролита.
Пример 5.1. Раствор, содержащий 33,2 г Ba(NO3)2 в 300 г воды, кипит при температуре 100,47 °С. Рассчитайте степень диссоциации соли a в растворе.
Решение:
По формуле (4.7) рассчитаем моляльность соли в растворе:
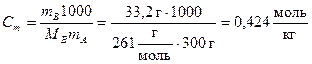 .
.
Из формулы (5.8) выразим изотонический коэффициент соли:
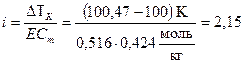 .
.
По формуле (5.11) рассчитаем степень диссоциации соли:
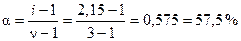 .
.
Ответ: a = 57,5 %.
Электролитическая диссоциация является обратимым процессом (исключение составляют сильные электролиты, в частности галогениды щелочных металлов NaCl, KBr и т. д.), поэтому важной количественной характеристикой состояния электролита в растворе является константа диссоциации КД (введена по аналогии с константой химического равновесия, см. §3.5). Так, для электролита Кn+Аn–, диссоциирующего в растворе на ионы по схеме:
| К n+ А n–«n+ Кz+ + n– Az –, | (5.12) |
выражение для КД (т. е. закон действующих масс) принимает вид:
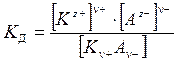 . . | (5.13) |
Если СМ – молярная концентрация электролита, а a – степень диссоциации, то равновесные концентрации частиц будут равны соответственно: [ Kz +] = a CМ; [ Az –] = a CМ; [ К n+ А n-] = = CМ (1 – a). Подставляя эти соотношения в уравнение (5.13) и принимая в простейшем случае n+ = n– = 1 (бинарный электролит), получаем:
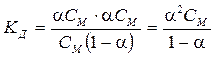 . . | (5.14) |
Уравнение (5.14) было выведено Оствальдом в 1888 г. и носит название «закон разбавления Оствальда». При небольших значениях a (для слабых электролитов) можно принять 1 – a» 1, тогда уравнение (5.14) переходит в:
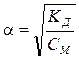 . . | (5.15) |
Как видно из уравнения (5.15), степень диссоциации обратно пропорциональна корню квадратному из концентрации электролита. При уменьшении концентрации электролита, например в 100 раз, степень диссоциации возрастает в 10 раз. КД зависит от природы растворителя и температуры, но не зависит от концентрации электролита в растворе.
Пример 5.2. Рассчитайте степень диссоциации уксусной кислоты CH3COOH в растворе молярной концентрации СМ = 0,1 М.
Решение:
По справочнику [Равдель] константа диссоциации CH3COOH КД = 1,754×10–5.
По формуле (5.15) степень диссоциации 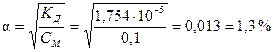 .
.
Таким образом, согласно принятой классификации (см. выше) уксусная кислота относится к слабым электролитам (a < 3 %).
Ответ: a = 1,3 %.
Уравнение закона разбавления (5.14) было получено для бинарного электролита. Для электролитов других типов оно имеет более сложный характер, но связывает те же переменные a и СМ. Тернарные и квартернарные электролиты диссоциируют ступенчато (в частности многоосновные кислоты – см. §1.3). Каждая ступень диссоциации характеризуется определённой константой диссоциации. Так, для ортофосфорной кислоты H3PO4 константы диссоциации каждой ступени при 25 °С равны [Равдель]:
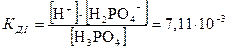 ,
,
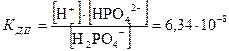 ,
,
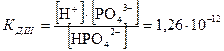 .
.
Из приведённого примера можно увидеть, что каждая последующая константа диссоциации меньше предыдущей примерно на 5 порядков. Соотношение КД,I > КД,II > КД,III характерно для всех многоосновных кислот. Причина этого в том, что первый ион водорода отрывается от молекулы кислоты легче, а последующие всё труднее, так как возрастает отрицательный заряд кислотного остатка.
Теория электролитической диссоциации Аррениуса – Оствальда позволила объяснить многие свойства разбавленных растворов слабых электролитов (например: реакции гидролиза (§5.6), значение концентрации водородных ионов (§5.3) и др.). На основе представлений об электролитической диссоциации были созданы теории электрической проводимости и диффузии в растворах электролитов, разработана осмотическая теория электродвижущей силы (ЭДС). Однако поведение концентрированных растворов слабых электролитов и растворов сильных электролитов количественно описать с позиций теории электролитической диссоциации оказалось невозможным. К недостаткам данной теории можно отнести следующие:
1. В растворах сильных электролитов нет динамического равновесия между молекулами и ионами. Многие экспериментальные данные указывают на почти полную диссоциацию сильных электролитов.
2. Пользуясь теорией электролитической диссоциации нельзя объяснить, почему разные методы определения КД для некоторых электролитов дают разные значения (например, методы, основанные на измерении электрической проводимости и ЭДС).
3. По теории электролитической диссоциации КД для данного электролита должна оставаться постоянной независимо от концентрации раствора. Однако только для очень слабых электролитов (NH4OH, CH3COOH) Кд остаётся при разбавлении более или менее постоянной. Для сильных электролитов (NaCl, MgSO4) она меняется в десятки раз. Остаётся необъяснимым и тот факт, что для некоторых электролитов при высоких концентрациях раствора a становится больше 1.
§5.2. Метод активностей. Элементы теории Дебая – Хюккеля
Теория электролитической диссоциации не учитывает силу электростатического взаимодействия Fe, возникающую между ионами в растворе электролита. Fe определяется по закону Кулона (открыт в 1785 г. Ш.О. де Кулоном):
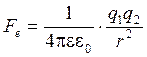 , , | (5.16) |
где q 1, q 2 – заряды взаимодействующих ионов, Кл;
r – расстояние между ионами;
e – относительная диэлектрическая проницаемость среды (растворителя);
e0 – диэлектрическая проницаемость вакуума, 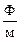 (8,85×10–12).
(8,85×10–12).
В растворах слабых электролитов влияние Fe на свойства раствора незначительно, что объясняется небольшой степенью диссоциации слабых электролитов. Однако, взаимодействия между ионами в растворах сильных электролитов настолько велики, что приводят к их связыванию. При этом, как следует из уравнения (5.16), чем больше концентрация электролита в растворе (т. е. чем меньше r между ионами), тем больше сила электростатического взаимодействия. При повышенных концентрациях в данных растворах может происходить ассоциация ионов. Так, в водных растворах установлено образование сложных ионов, например BaCl+, AgCl2–, LiCl2–. При разбавлении раствора эти частицы диссоциируют. Поэтому, с повышением концентрации сильных электролитов в растворе даже при их полной диссоциации свойства раствора меняются в направлении, аналогичном уменьшению степени диссоциации слабых электролитов. Таким образом, особые свойства растворов сильных электролитов, необъяснимые с позиций теории электролитической диссоциации, обусловлены наличием между ионами в растворе значительных электростатических сил.
Для растворов сильных электролитов (а также концентрированных растворов слабых электролитов) используют метод активностей, разработанный в 1907 г. Г.Н. Льюисом. В основе метода лежит понятие «активность», т. е. кажущаяся, или эффективная концентрация. Активность a – это условная концентрация, при использовании которой законы, описывающие растворы, имеют одинаковую форму, независимо от концентрации.
Рассмотрим сильный электролит К n+ А n–, полностью диссоциирующий на ионы по схеме (5.12). В этом случае знак обратимой реакции меняется на знак необратимой реакции:
| К n+ А n–® n+ Кz+ +n– Az–; n+ + n– = n. | (5.17) |
Активность катионов Кz +будет равна а +, активность анионов Az– – равна а–. Однако определить активности отдельных ионов из опытных данных невозможно. Это связано с тем, что в растворах электролитов одновременно присутствуют и катионы и анионы (получить раствор, содержащий только катионы или только анионы, нельзя). Поэтому для растворов электролитов вводится понятие «средняя ионная активность» (обозначается а ±), представляющая среднее геометрическое из активностей ионов, составляющих данный электролит:
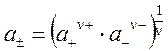 . . | (5.18) |
Общая активность электролита а связана со средней ионной активностью a ± соотношением:
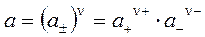 . . | (5.19) |
Существует три способа выражения средней ионной активности а ±– через среднюю ионную моляльность Cm ,±, среднюю ионную молярность CM ,±, среднюю ионную молярную долю N ±:
| a ±, m = g±, m × Cm ,±, | (5.20) | |
| a ±, М = g±, М × CM ,±, | (5.21) | |
| a ±, N = g±, N × N ±, | (5.22) |
где g±, m – моляльный средний коэффициент активности;
g± ,М – молярный средний коэффициент активности;
g±, N – рациональный средний коэффициент активности.
Коэффициенты активности g±, m и g±, М называют также практическими средними коэффициентами активности электролита. В термодинамике растворов электролитов обычно используется моляльная шкала концентраций 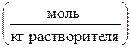 , преимущество которой заключается в том, что добавление в раствор другого вещества не изменяет моляльности данного вещества [Пригожин, 2009; с. 214]. Поэтому в дальнейшем будем пользоваться средней ионной активностью a ±, m и моляльным средним коэффициентом активности g±, m, обозначая их для краткости a± и g±.
, преимущество которой заключается в том, что добавление в раствор другого вещества не изменяет моляльности данного вещества [Пригожин, 2009; с. 214]. Поэтому в дальнейшем будем пользоваться средней ионной активностью a ±, m и моляльным средним коэффициентом активности g±, m, обозначая их для краткости a± и g±.
Средний коэффициент активности электролита g± является средним геометрическим из коэффициентов активности катиона g+ и аниона g-:
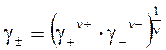 . . | (5.23) |
Средняя ионная моляльность Cm ,± является средним геометрическим из моляльностей катиона Cm ,+ и аниона Cm ,-:
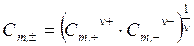 . . | (5.24) |
Моляльности катионаи аниона определяются из моляльности электролита Cm:
| Cm ,+ = n+× Cm и Cm ,- = n- × Cm. | (5.25) |
Подставляя значения Cm ,+ и Cm ,- из уравнений (5.25) в уравнение (5.24), получим:
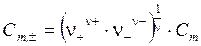 . . | (5.26) |
У бинарного 1,1-зарядного электролита (NaCl, KNO3 и др.) n+ = n- = 1, и 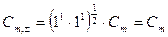 . Такой же результат получим для бинарного 2,2-зарядного электролита (MgSO4, CaSO4 и др.). Для электролита типа К2А3 (например, Al2(SO4)3)
. Такой же результат получим для бинарного 2,2-зарядного электролита (MgSO4, CaSO4 и др.). Для электролита типа К2А3 (например, Al2(SO4)3) 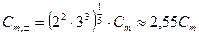 . Таким образом, в общем случае средняя моляльность ионов электролита Cm ,± не равна моляльности электролита Cm.
. Таким образом, в общем случае средняя моляльность ионов электролита Cm ,± не равна моляльности электролита Cm.
Экспериментальные значения среднего коэффициента активности g± можно получить из эбуллиоскопических, криоскопических и осмометрических измерений (см. § 4.3), или по электрохимическим данным. После этого по формулам (5.20 – 5.22) можно рассчитать а ±. Средний коэффициент активности зависит от зарядов и концентрации всех ионов в растворе. Для количественной характеристики влияния этих факторов используется правило ионной силы (Льюис, 1907 г.). Ионной силой I раствора сильного электролита или смеси сильных электролитов называется полусумма произведений моляльностей каждого иона Cmi на квадрат его заряда zi:
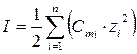 . . | (5.27) |
Согласно правилу ионной силы, в разбавленных растворах средний коэффициент активности электролита зависит только от ионной силы раствора и не зависит от природы других ионов, находящихся в растворе.
Это правило справедливо при концентрации раствора менее 0,01 – 0,02 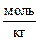 , но приближённо им можно пользоваться до концентрации 0,1 – 0,2 .
, но приближённо им можно пользоваться до концентрации 0,1 – 0,2 .
Подстановка активности вместо концентрации в уравнения, действующие для разбавленных растворов слабых электролитов, делает их применимыми для любых рассматриваемых растворов электролитов. Так, например, для концентрированного раствора слабого электролита или для раствора сильного электролита, диссоциирующего по схеме (5.12), константа диссоциации может быть рассчитана по формуле:
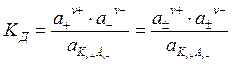 . . | (5.28) |
Введение понятия об активности не раскрывает механизм процессов взаимодействия ионов между собой и с молекулами растворителя. Это лишь удобный приём, позволяющий определить свойства многих растворов. Зависимость g± (а следовательно и а ±) от концентрации может быть объяснена в рамках электростатической теории разбавленных растворов сильных электролитов Дебая – Хюккеля. Данная теория была разработана в 1923 г. П. Дебаем и Э. Хюккелем.
В основе теории лежит идея о наличии вокруг каждого иона в растворе ионной атмосферы. Если мысленно выделить в разбавленном растворе сильного электролита один центральный ион (например, катион), то ионы противоположного знака (анионы) будут чаще наблюдаться около него, чем ионы с одноимённым зарядом. Такое статистическое распределение ионов вокруг выбранного центрального иона будет устанавливаться под влиянием двух факторов:
1. электростатических сил притяжения и отталкивания, которые стремятся расположить ионы упорядоченно, как в кристаллической решётке;
2. теплового движения ионов, под влиянием которого ионы стремятся расположиться хаотически.
В результате вокруг центрального иона устанавливается некоторое промежуточное статистическое распределение ионов, так называемая ионная атмосфера (рис. 5.4). При этом вокруг центрального иона будет некоторая избыточная плотность зарядов противоположного знака, которая по мере удаления от центрального иона убывает и на бесконечно большом расстоянии стремится к нулю. Фактически уже на расстоянии несколько нм от центрального иона величина этого избыточного заряда становится очень малой и может считаться практически равной нулю. Весь избыточный заряд ионной атмосферы равен по модулю и противоположен по знаку заряду центрального иона. Для упрощения математических расчётов Дебай и Хюккель предположили, что избыточный заряд ионной атмосферы «размазан» в пространстве вокруг центрального иона с убывающей плотностью.
Авторы теории сделали ряд допущений, справедливых только для предельно разбавленных растворов:
1. Электролит в растворе диссоциирует полностью, и концентрацию ионов рассчитывают по аналитической концентрации электролита.
2. Ионы рассматриваются в виде математических точек; такое допущение возможно только в разбавленных растворах, когда можно пренебречь собственным объёмом ионов.
3. Учитывается только кулоновское ионное взаимодействие (см. уравнение (5.16)) и игнорируются все другие виды взаимодействий (например, ион-дипольное взаимодействие, образование ассоциированных комплексов и т. д.).
4. Не учитывается изменение диэлектрической проницаемости раствора по сравнению с диэлектрической проницаемостью чистого растворителя.
5. Распределение ионов в ионной атмосфере подчиняется классической статистике Максвелла – Больцмана (см. рис.3.3), а сама ионная атмосфера рассматривается как непрерывная среда.
6. Электростатическое взаимодействие рассматривается как взаимодействие между центральным ионом и его ионной атмосферой.
Таким образом, в теории Дебая – Хюккеля остаётся неучтённым ряд существенных взаимодействий и свойств ионов. Теория не учитывает, например, сольватацию ионов (см. §5.1), особенности строения ионов, их поляризуемость и т. д., что существенно ограничивает применимость теории.
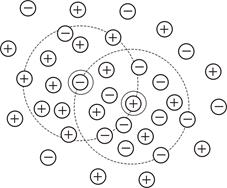 | Рис. 5.4. Схема образования ионной атмосферы вокруг центрального иона в растворе сильного электролита |
Электростатическая теория сильных электролитов позволяет вычислить средний коэффициент активности g± сильного электролита. Для этого используют уравнение, выведенное Дебаем и Хюккелем, и носящее название «предельный закон Дебая – Хюккеля». В первом приближении предельный закон выглядит следующим образом:
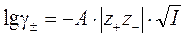 , , | (5.29) |
где z +, z – – заряды катиона и аниона;
I – ионная сила.
Коэффициент А рассчитывают по формуле:
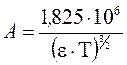 , , | (5.30) |
где: e – относительная диэлектрическая проницаемость растворителя при температуре Т.
Пример 5.3. Рассчитайте ионную силу раствора I, средний коэффициент активности g± и среднюю ионную активность a ± K2SO4 концентрацией Cm = 0,01 , растворённого в воде вместе с NaCl (0,05 ) и H2SO4 (0,1 ) при температуре 50 °С.
Решение:
По формуле (5.27) рассчитаем ионную силу раствора:
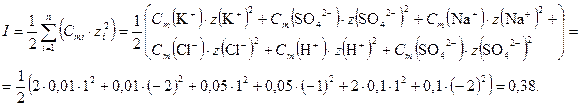
При T = (50 + 273,15) = 323,15» 323 К относительная диэлектрическая проницаемость воды e = 69,73 [Равдель].
По формуле (5.29) 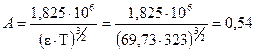 .
.
По предельному закону Дебая – Хюккеля (5.28) рассчитаем средний коэффициент активности K2SO4: 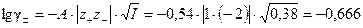 , отсюда
, отсюда 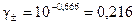 .
.
По формуле (5.26) рассчитаем среднюю ионную моляльность K2SO4:
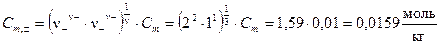 .
.
По формуле (5.20) рассчитаем среднюю ионную активность K2SO4:
a ± = g± × Cm ,± = 0,216 × 0,0159 = 0,00324 .
Ответ: I = 0,38; g± = 0,216; a ± = 0,00324 .
Из уравнения (5.28) следует, что при постоянной температуре Т зависимость lgg± от 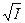 выражается прямой линией с угловым коэффициентом – А ô z + z –ô. В действительности это справедливо только для очень разбавленных растворов электролитов. Так, для 1,1-зарядных электролитов зависимость (5.28) соблюдается в растворах с ионной силой I порядка 10–2 и меньше, т. е. при Сm = 0,01 и в более разбавленных растворах. До сих пор ещё не существует достаточно строгой теории, которая бы позволила рассчитать в согласии с опытом зависимость среднего коэффициента активности электролита от концентрации для концентрированных растворов.
выражается прямой линией с угловым коэффициентом – А ô z + z –ô. В действительности это справедливо только для очень разбавленных растворов электролитов. Так, для 1,1-зарядных электролитов зависимость (5.28) соблюдается в растворах с ионной силой I порядка 10–2 и меньше, т. е. при Сm = 0,01 и в более разбавленных растворах. До сих пор ещё не существует достаточно строгой теории, которая бы позволила рассчитать в согласии с опытом зависимость среднего коэффициента активности электролита от концентрации для концентрированных растворов.

