2015-09-06
2015-09-06 884
884Стадии гипоксии коррелируют с фазными изменениями содержания ATФ и ведущих энергозависимых процессов в клетках. Только на последних этапах кислородного голодания уровень энергетического дефицита становится достаточным для запуска основных механизмов, приводящих к нарушению жизнедеятельности и гибели клетки. Стремительное увеличение концентрации аденозинмонофосфата (AMФ) сопровождается активацией протеинкиназной системы, что является дополнительным механизмом разрушения клеточных мембран.
Гликолиз не предотвращает снижения уровня ATФ на поздних стадиях кислородного голодания. Однако, увеличение утилизации GL астроцитами вследствие глутаматиндуцированной активации Na+/K+-ATФ-азы приводит к ее метаболизации по гликолитическому пути до лактата, который, высвобождаясь, трансформируется нейронами в пируват и используется как адекватный энергетический субстрат. В течение нескольких минут после начала острой фокальной церебральной ишемии развивается дефицит макроэргов (ATФ, креатинфосфата) в ткани мозга.
Особенности энергетических изменений в ткани мозга зависят и от локализации ишемического процесса. В большинстве областей мозга реперфузия сопровождается полным или частичным возвращением энергетического метаболизма к нормальным показателям: увеличиваются концентрации ATФ и креатинфосфата, снижается уровень лактата. В то же время в селективно чувствительных к ишемии нейронах (CA1 зона гиппокампа, дорсолатеральный отдел стриатума) изменения энергетического метаболизма имеют двухфазный характер: вслед за кратковременной нормализацией отмечается его вторичное торможение.
Развитие «постишемической гипоксии» приводит к значительному нарушению функций митохондрий, уменьшению производства никотинамиддинуклеотидфосфата (NADP) в синаптосомах, что оказывает дополнительное воздействие на процессы необратимого повреждения ткани мозга.
Нейроны, подверженные тяжелой ишемии (мозговой кровоток менее 10-15 мл/100 г в 1 мин), а следовательно, и быстро развивающимся грубым нарушениям энергетического метаболизма, не могут поддерживать ионный градиент мембран за счет «обесточивания» Na+/K+-ATФ-aзной ферментной системы. Скорость развития аноксической деполяризации мембран нейронов зависит от глубины и длительности ишемии и ведет к некротической смерти клетки. Вероятно, энергетический дефицит является главенствующим механизмом гибели нейронов в области центрального инфаркта (ядерной зоне ишемии). В зоне пенумбры более «мягкая» ишемия инициирует развитие комплекса биохимических преобразований, поддерживаемых реакцией генома и молекулярными последствиями ишемического процесса: включением генов раннего реагирования с вторичной экспрессией генов, кодирующих цитокины, молекулы адгезии, другие провоспалительные и трофические факторы, а также гены апоптоза.
Энергетический дефицит и лактат-ацидоз являются триггерами каскада патобиохимических реакций, протекающих во всех основных клеточных пулах ЦНС и приводящих к формированию инфаркта мозга по двум основным механизмам: некроза и апоптоза.
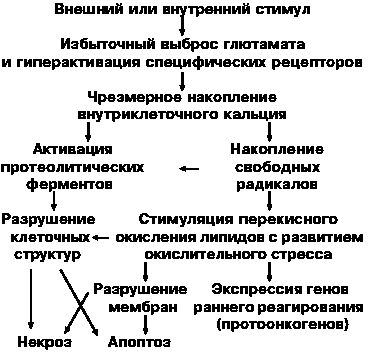
Рисунок 5. Схема этапов «ишемического каскада»