 2015-09-06
2015-09-06 3328
3328Рассмотрим два метода определения диссоциации молекулы – аналитический и графический. При аналитическом способе нахождения энергии диссоциации необходимо анализировать двучленную формулу колебательной энергии:
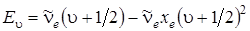
и попытаться экстраполировать ее к границе диссоциации, соответствующей схождению колебательных уровней. Граница диссоциации наступит в том случае и при таких u, когда разность энергий между соседними уровнями будет равна 0. Как видно в (3.3), колебательные интервалы линейно зависят от u (если не учитывать вторые ангармоничности) и стремятся к нулю при приближении u к своему максимальному значению umaх. Возьмем разность между соседними колебательными уровнями у границы диссоциации и приравняем ее к нулю. Из полученного соотношения найдем umaх. Итак,
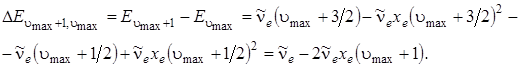 (3.41)
(3.41)
Приравняв это выражение нулю, получим 1 – 2 хe (umax + 1) = 0, откуда umax = 1/2 хe (учитывается тот факт, что хe << 1).
Подставляя umax в выражение для колебательной энергии, получим значение энергии диссоциации молекулы:
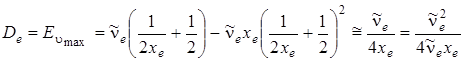 . (3.42)
. (3.42)
Необходимо отметить, что полученная формула весьма приближенная и дает несколько завышенные значения (примерно 17 %) энергии диссоциации. Формулу (3.42) легко получить и несколько иначе.
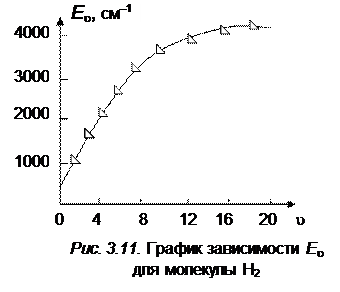
У границы диссоциации существует umax, после которого следующий дискретный уровень с umax + 1 будет отсутствовать. Граница диссоциации получается при таком значении u, при котором E u как функция u достигает максимума. Если построить график зависимости E u от u (рис. 3.11), то функция E будет нелинейной и у границы диссоциации при umax + 1 она должна была бы повернуть вниз, если бы такое umax + 1 существовало (за счет существенного влияния квадратичного члена).
Максимум функции E u соответствует условию
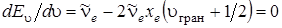 . (3.43)
. (3.43)
Из последнего равенства получим
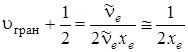 . (3.44)
. (3.44)
Подстановка uгран в формулу (3.42) дает значение De
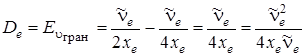 . (3.45)
. (3.45)
Расчетные значения энергии диссоциации не совпадают с экспериментально полученными значениями D 0, которые меньше 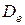 на величину нулевой энергии молекулы. Действительно, как видно из рисунка 3.7,
на величину нулевой энергии молекулы. Действительно, как видно из рисунка 3.7,
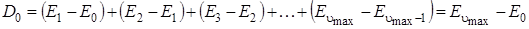 ,
,
так как все внутренние значения энергии уничтожаются. Так как
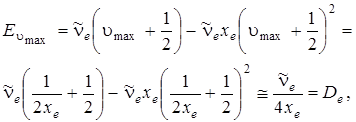
то
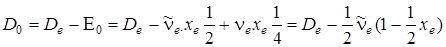 . (3.46)
. (3.46)
Непосредственно из опыта определяется D 0, а значение находится по формуле (3.45), если известны 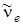 и хe.
и хe.
Если энергию отсчитывать от u = 0, а не от минимума кривой потенциальной энергии, то двучленную формулу (3.29) можно записать в виде:
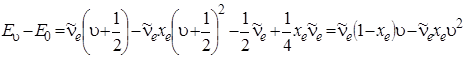 . (3.47)
. (3.47)
Если положить в этой формуле, 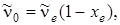 а
а 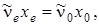 то равенство (3.47) можно переписать следующим образом:
то равенство (3.47) можно переписать следующим образом:
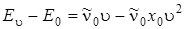 , (3.48)
, (3.48)
где частота 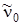 и постоянная ангармоничности х 0 соответствуют уровню u = 0.
и постоянная ангармоничности х 0 соответствуют уровню u = 0.
Энергию диссоциации D 0 находим так, как и раньше. Определим umax, при котором функция E u – E 0 максимальна. Взяв производную по u от E u – E 0 и приравняв ее нулю, получим
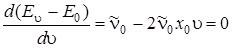 ,
,
откуда
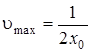 .
.
Подставив полученное значение в (3.48), получим
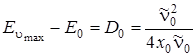 . (3.49)
. (3.49)
Частоты переходов между соседними уровнями энергии на основании равенства (3.48) определим по формуле:
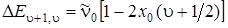 . (3.50)
. (3.50)
Сумма первых разностей по всем колебательным квантовым числам до umax, равна энергии диссоциации D 0. Введение коэффициента u + 1/2, а не u в первые разности связано с определением энергии диссоциации.
3.4.2. Метод графической экстраполяции
(метод Берджа – Шпонер)
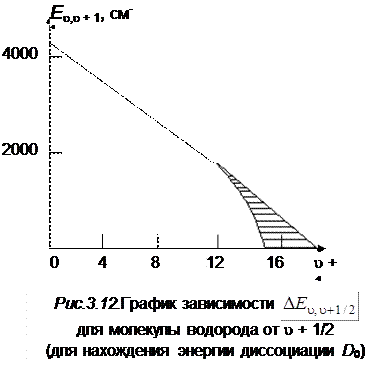
Сущность метода графической экстраполяции заключается в том, что строится график зависимости колебательных интервалов D E u + 1 , u, от u + 1, который экстраполируется до пересечения с осью абсцисс. Площадь под этой кривой соответствует энергии диссоциации De. Заштрихованная область дает нижний предел . На рис. 3.12 приведен график этой зависимости для молекулы водорода.
При справедливости двучленной формулы для колебательной энергии (3.30), график является прямой линией и соответствующая площадь выражается формулой
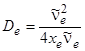 . (3.51)
. (3.51)
При небольших отступлениях от линейности получаются несколько иные значения диссоциации. Действительно, при линейной зависимости первой разности  ордината в нулевой точке (u + 1), т. е. при u + 1=0, равна . В точке, для которой ордината обращается в нуль, значение абсциссы
ордината в нулевой точке (u + 1), т. е. при u + 1=0, равна . В точке, для которой ордината обращается в нуль, значение абсциссы 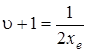 , как это следует из (3.41). Площадь треугольника определяется формулой
, как это следует из (3.41). Площадь треугольника определяется формулой 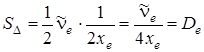 , что согласуется с (3.42). Здесь энергию диссоциации отсчитывают от минимума потенциальной энергии. Реальную же энергию диссоциации D 0 следует отсчитывать от уровня u = 0, для которого нулевая энергия равна
, что согласуется с (3.42). Здесь энергию диссоциации отсчитывают от минимума потенциальной энергии. Реальную же энергию диссоциации D 0 следует отсчитывать от уровня u = 0, для которого нулевая энергия равна
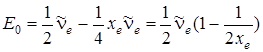 ,
,
а энергия диссоциации
D 0 = De – E 0 . (3.52)
Частоты переходов между соседними уровнями, как следует из (3.50), равны
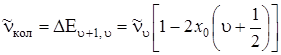 , (3.53)
, (3.53)
где отличается от на величину ангармоничности 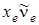 . Следовательно
. Следовательно 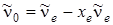 , т. е. величина практически совпадает с экспериментально измеряемой частотой
, т. е. величина практически совпадает с экспериментально измеряемой частотой
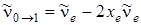 . (3.54)
. (3.54)
При графическом определении энергии диссоциации D 0 нужно откладывать по оси абсцисс u + 1/2, а по оси ординат 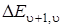 . Из формулы (3.50) получаем прежний график (прямую линию), что на рис. 3.12 только смещенный вправо на 1/2 (см. рис. 3.13).
. Из формулы (3.50) получаем прежний график (прямую линию), что на рис. 3.12 только смещенный вправо на 1/2 (см. рис. 3.13).

Разница значений и D 0 выражается площадью, заштрихованной на рис. 3.13. Оси абсцисс на рис. 3.13 совпадают. Действительно, при u + 1, согласно (3.35) 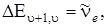 а при
а при 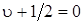 согласно (3.50)
согласно (3.50) 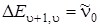 . Заштрихованная площадь равна
. Заштрихованная площадь равна
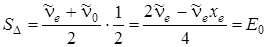 (нулевая энергия).
(нулевая энергия).
Здесь учтено, что . Следовательно, площадь треугольника S Dв координатах и 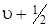 меньше площади другого треугольника на величину E 0. Итак, D 0 = De – E 0 или же
меньше площади другого треугольника на величину E 0. Итак, D 0 = De – E 0 или же
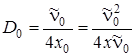 .
.
 |
Получаемые значения энергии диссоциации
и D 0, рассчитываемые описанными выше методами, отличаются от истинных примерно на 15 %, что указывает на то, что формула Морзе, аппроксимирующая реальную кривую потенциальной энергии, является лишь грубым приближением. Экспериментальные точки ложатся не на прямую, а на кривую, которая загибается раньше, т. е. пересекает ось абсцисс при меньших значениях u, чем это следует из анализа двучленной формулы (3.30). На рис. 3.12 и 3.14 показаны кривые зависимости D E u+1,uот квантового числа u для молекул H2 и Li2. 
Энергия диссоциации определяется как площадь, ограниченная осями координат и реальной кривой, измеренная в соответствующем масштабе.
Энергию диссоциации можно измерить не только спектроскопическими, но и другими методами, которые мы здесь не рассматриваем (например, методом электронного удара, термическим или термохимическим. Знание энергий диссоциации молекул необходимо для термодинамических расчетов в химических реакциях.
Следует отметить, что задача определения энергии диссоциации по спектрам не проста, как может показаться из приведенного изложения. Требуется полная и точная расшифровка колебательного спектра, что не всегда можно довольно полно провести. Например, молекулы Н2, N2 и О2 при обычных условиях не поглощают ИК-радиации (у них отсутствует дипольный момент). Колебательные частоты у аналогичных молекул получают из спектров КР или из ИК-спектров при высоких давлениях, когда при соударениях симметрия (D ¥ h) молекулы нарушается и могут наблюдаться оптически неактивные полосы.
3.5. Колебательные спектры двухатомных
молекул с изотопным замещением
Как мы видели в 2.2, положение колебательных полос в ИК-спектрах молекул зависит от приведенной массы m молекулы. Если один из атомов в молекуле заменить ее изотопом, то это почти не повлияет на ее электронную оболочку и, следовательно, на межатомные расстояния и квазиупругую постоянную k, но существенно изменит приведенную массу 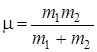 .
.
Так как положение колебательных полос обратно пропорционально приведенной массе m, то при более тяжелой изотопной молекуле положение полос будет сдвигаться влево в частотной шкале, а величина сдвига будет определяться различием в приведенных массах. например, для водорода Н2 и D2 это различие максимально, так как приведенные массы различаются в 2 раза. для других более тяжелых молекул это различие невелико.
Рассмотрим изменение колебательных частот для молекул, содержащих атом водорода Н, который может быть заменен атомом дейтерия D, имеющим вдвое большую массу. Указанная процедура в химии называется дейтерированием, которое приводит к довольно значительному изменению частот колебаний. Например, возьмем отношение частот колебаний Н2 и D2
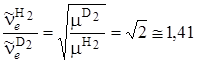 , (3.55)
, (3.55)
т. е. частота колебаний молекулы водорода в 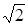 раз больше частоты колебаний молекулы дейтерия. Действительно, сравнение частот колебаний этих молекул, приведенных в табл. 3.1 показывает, что их отношение равно 1,41, что совпадает с (3.55). Если сравнить различие частот колебаний в молекуле
раз больше частоты колебаний молекулы дейтерия. Действительно, сравнение частот колебаний этих молекул, приведенных в табл. 3.1 показывает, что их отношение равно 1,41, что совпадает с (3.55). Если сравнить различие частот колебаний в молекуле 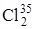 и
и 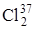 ,то оно будет небольшим, так как отношение частот колебаний близко к единице:
,то оно будет небольшим, так как отношение частот колебаний близко к единице:
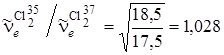 . (3.56)
. (3.56)
При частоте колебаний 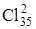 , равной 560 см–1, это различие составит 16 см–1, а разница в частотах для водорода (
, равной 560 см–1, это различие составит 16 см–1, а разница в частотах для водорода (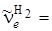 4401 см–1) и дейтерия (
4401 см–1) и дейтерия (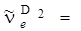 3118 см–1) составляет 1283 см–1.
3118 см–1) составляет 1283 см–1.
Рассмотрим в общем случае более подробно отношение изотопических частот r. обозначим массы атомов в изотопически незамещенной молекуле через m 1 и m 2, а в изотопически замещенной – через 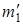 и
и 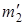 . тогда выражение
. тогда выражение
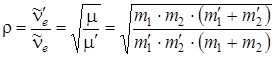 . (3.57)
. (3.57)
в случае изотопического замещения лишь одного из атомов (например, m 2 = m' 2) сводится к формуле:
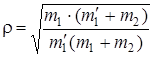 . (3.58)
. (3.58)
При дейтерировании двухатомной молекулы соотношение (3.58) можно переписать следующим образом:
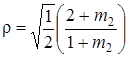 , (3.59)
, (3.59)
где m 2 – масса атома, отличного от водорода. Когда эта масса достаточно велика, т. е. m 2 >> m 1, то величина r будет стремиться к своему предельному значению 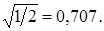 Этот результат физически понятен, так как при большой массе m 2 практически колеблется только атом водорода (m 1).
Этот результат физически понятен, так как при большой массе m 2 практически колеблется только атом водорода (m 1).
Для случая, когда массы изотопов будут мало отличаться друг от друга (например, ( и ), изменение частот колебаний будет невелико, т. е. rбудет мало отличаться от единицы. Пусть m 1 = m 2 и m '1 = m '2, но m '2 = m 1 + D, где D есть разность в изотопических массах, на основании (3.57) получим
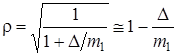 , (3.60)
, (3.60)
т. е. относительное изменение частот порядка 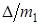 . Для указанных молекул это отличие равно 0,057. Необходимо при этом отметить, что различие в частотах колебаний изотопных молекул в обертонах выше, чем у основных тонов, и это различие тем больше, чем выше порядок обертона.
. Для указанных молекул это отличие равно 0,057. Необходимо при этом отметить, что различие в частотах колебаний изотопных молекул в обертонах выше, чем у основных тонов, и это различие тем больше, чем выше порядок обертона.
Метод изотопических замещений оказывается очень полезным при рассмотрении колебаний многоатомных молекул, так как позволяет относить полученные в ИК-спектре полосы к колебаниям соответствующих групп атомов в сложной молекуле.
В заключение необходимо отметить, что изотопическое замещение одного из атомов в двухатомной молекуле может приводить к появлению полос в ИК-спектре поглощения для молекул, не обладающих дипольным моментом и относящихся к симметрии D ¥ h. Если молекула состоит из разных изотопов (например, HD и Сl37 Cl35), то колебание у этой молекулы происходит несимметричным образом и возникает переменный дипольный момент, обуславливающий поглощение ИК радиации. Симметрия молекулы при этом нарушается колебаниями, которые не являются полносимметричными. Только при одинаковых ядрах двухатомная молекула при колебаниях сохраняет симметрию D ¥ h, при этом указанная молекула не будет поглощать или испускать ИК-радиацию. Чем больше различие в массах колеблющихся атомов, тем больше асимметрия колебаний, тем интенсивнее соответствующая полоса в ИК-спектре.
Эффект изотопного замещения в колебательных спектрах дает чувствительный и довольно точный метод обнаружения изотопов и измерения их относительных количеств путем измерения интенсивностей линий.
3.6. Колебательно-вращательные
спектры двухатомных молекул
3.6.1. Общая характеристика колебательно-вращательных
спектров двухатомной молекулы.
Величины колебательных квантов (100 – 1000 см –1) на два порядка больше величин вращательных квантов (1 – 10 см–1). Поэтому возбуждение колебательных уровней энергии всегда сопровождается изменением вращательной энергии молекулы, что приводит к возникновению колебательно-вращательного спектра, т. е. происходит одновременное возбуждение как колебаний, так и вращений молекулы. Так как изменения вращательной энергии малы по сравнению с изменениями колебательной энергии, то в спектрах получаются полосы, которые соответствуют определенному колебательному переходу и структура которых обусловлена вращательными переходами т. е. колебательно-вращательные полосы. Вращательную структуру колебательных полос можно разрешить, применяя приборы с высокой разрешающей способностью (большая дисперсия) и при записи спектров газовых образцов. В этом случае вращательные полосы распадаются на ряд отдельных линий, соответствующих отдельным вращательным переходам.
Изменение внутренней энергии молекулы D Е вн можно представить как изменение ее колебательной D Е кол, и вращательной D Е вр энергий. Пусть колебательный переход происходит между нижним и верхним колебательными уровнями, а внутренняя энергия нижнего состояния будет представляться суммой колебательной и вращательной энергий:
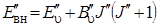 . (3.61)
. (3.61)
Аналогичную формулу можем записать и для верхнего колебательного уровня
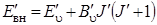 . (3.62)
. (3.62)
В формулах (3.61) и (3.62) значок «''» относится к нижнему, а «'» – к верхнему колебательному уровню.Связь колебаний с вращениями выражается в том, что вращательная постоянная В u оказывается зависящей от колебательного квантового числа u. Эта зависимость приближенно выражается формулой
B u =Be – a e (u + 1/2), (3.63)
где a e – постоянная, отношение которой к Be имеет такой же порядок, как и постоянная ангармоничности xe, т. е. не превышает нескольких сотых. Be – вращательная постоянная, соответствующая равновесному расстоянию между атомами.
Зависимость вращательной постоянной от колебательного (u) квантового числа обусловлена изменением момента инерции молекулы при возбуждении колебаний. Действительно, при возбуждении высоких колебательных уровней двухатомной молекулы ее равновесное расстояние увеличивается, что приводит к увеличению вращательного момента (I) и соответственно к уменьшению вращательной постоянной B. Например, для молекулы СО значения вращательных постоянных для u = 0 и u = 1 соответственно равны B 0 = 1,915 см–1 и В 1 = 1,898 см–1, что соответствует величине a е = 0,0175 см –1 (отношение a/ В = 0,009, постоянная ангармоничности xe для этой молекулы равна 0,0061).
Для выяснения общего характера вращательной структуры колебательного спектра мы воспользуемся независимостью колебательной и вращательной энергии молекулы в предположении жесткости вращения молекулы. Вычислим разность значений энергии между двумя колебательно-вращательными уровнями
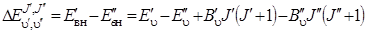 . (3.64)
. (3.64)
Если измерять энергию в см–1, то эта разность даст нам частоту чисто колебательного перехода 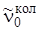 , которая получилась бы при J' = J'' = 0, и частоты вращательных переходов
, которая получилась бы при J' = J'' = 0, и частоты вращательных переходов
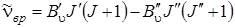 . (3.65)
. (3.65)
В силу запрета переходов J' = J'' = 0 (даже при правиле отбора D J = 0) нулевая линия чисто колебательного перехода не может наблюдаться, и ее положениe определяется расчетом. Вращательная структура колебательной полосы определяется выражением (3.65). Существенная разница выражения (3.65) по сравнению с аналогичным выражением (2.17) для чисто вращательных спектров заключается в том, что в выражении (3.65) В' u и В' u относятся к разным колебательным состояниям молекулы, в то время как в чисто вращательных спектрах постоянная В относилась к одному колебательному состоянию. Влияние центробежного растяжения на величину B в чисто вращательных спектрах для небольших J мало, и им будем пренебрегать. Отличие В' u от В' u в формуле (3.65), определяемой зависимостью вращательной постоянной от колебательного квантового числа u, более существенно. Согласно правилам отбора по вращательному квантовому числу J могут комбинировать между собой соседние вращательные уровни, относящиеся к различным колебательным состояниям, одинаковые вращательные уровни (кроме уровней J'–J" = 0) и вращательные уровни через один, т.е., D J = J' – J" = 0, ±1, ±2. Заданному значению D J будет соответствовать ряд переходов, отличающихся J' и соответственно J". Совокупность вращательных линий при таких переходах называют ветвью соответствующей полосы. Эти ветви принято обозначать латинскими буквами в зависимости от D J:
D J = J' – J" = –2 (О -ветвь)
D J = J' – J" = –1 (Р -ветвь)
D J = J' – J" = 0 (Q -ветвь)
D J = J' – J" = +1 (R -ветвь)
D J = J' – J" = +2 (S- ветвь)
Одни из таких ветвей будут обнаруживаться в спектрах ИК-поглощения и испускания, другие – в спектрах КР. Для всех этих переходов с изменением дипольного момента D J будет изменяться на ±1, т. е. будут обнаруживаться R и Р -ветви (иногда может обнаруживаться Q -ветвь, если в нормальном состоянии двухатомная молекула будет обладать электронным орбитальным моментом, отличным от нуля). В спектрах КР наблюдаются Q. -, S - и О -ветви. Рассмотрим характеристики ветвей для ИК-поглощения.
3.6.2 Колебательно - вращательные спектры. ИК - поглощения
Колебательно-вращательные спектры, соответствующие дипольным переходам в ИК области, подвержены следующим правилам отбора для вращательного квантового числа
D J = J' – J" = ±1. (3.66)
Это правило отбора относится к молекулам, не обладающим в нижнем электронном состоянии механическим моментом (преимущественно сюда относятся молекулы, находящиеся в S-состоянии). Колебательные полосы таких молекул будут состоять из R-ветви, т. е. совокупности линий, для которых DJ = J' – J" = 1, и Р-ветви, т. е. совокупности линий, для которых DJ = J' – J" = – 1.
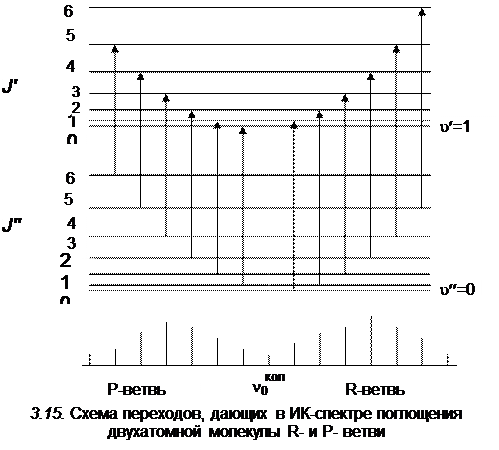
Схема переходов, дающих колебательно–вращательную полосу для R- и P-ветвей в ИК-спектре поглощения, показана на рис. 3.15. Частоты R-ветви всегда расположены в коротковолновую сторону от частоты 
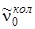 , а частоты Р-ветви расположены с длинноволновой стороны.
, а частоты Р-ветви расположены с длинноволновой стороны.
Выражая D Е = Е 'вн – Е' 'вн и частоты переходов в одних и тех же единицах (см–1), мы получим соответствующие формулы для R -ветви
(J'– J" = 1)
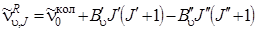 , (3.67)
, (3.67)
или, выражая J'' через J' (J'' = J' – 1), получим
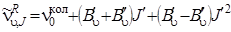 , (3.68)
, (3.68)
где J' = 1, 2, 3,...(см. рис.3.15)
Аналогично получим, если произвести замену J' через J' '(J'= J'' + 1)
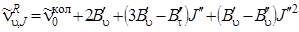 , (3.69)
, (3.69)
где J' = 0, 1, 2, 3,...
Формулы (3.68) и (3.69), дают ряд линий с частотами 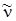 >
>  которые при B'u = B ''u, были бы равноотстоящими, но в силу наличия квадратичного члена с B' u< B' u будут сходиться. Если бы B' u > B' 'u, линии расходились бы. Для колебательно-вращательных спектров имеет место именно первый случай B' u < B'' u, так как u' > u''.
которые при B'u = B ''u, были бы равноотстоящими, но в силу наличия квадратичного члена с B' u< B' u будут сходиться. Если бы B' u > B' 'u, линии расходились бы. Для колебательно-вращательных спектров имеет место именно первый случай B' u < B'' u, так как u' > u''.
Аналогичную картину распределения линий вращательной структуры имеем для Р -ветви (J' – J'' = –1).
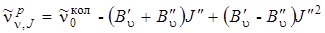 , (3.70)
, (3.70)
если выражать J' через J'' (здесь J' ' = 1,2, 3,...). Можно получить и другую формулу
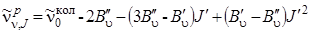 , (3.71)
, (3.71)
если; выразить J'' через J'. Формулы (3.70) и (3.71) также дают ряд линий с 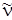 <
< 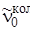 , которые при B' u = B'' u были бы равноотстоящими, а в силу наличия квадратичного члена с B' u < B'' uбудут расходиться (при B' u > B'' u они сходились бы). Проанализируем формулы (3.68) и (3.70), которые подобны по своей записи, если J' и J'' принимают значения 1, 2, 3,... Разница этих формул состоит только в знаке линейного члена. Если в формуле (3.68) принять J' = m, где m = 1, 2, 3,..., а в равенстве (3.70) положить J'' = –m, то обе формулы становятся одинаковыми и их можно записать в виде
, которые при B' u = B'' u были бы равноотстоящими, а в силу наличия квадратичного члена с B' u < B'' uбудут расходиться (при B' u > B'' u они сходились бы). Проанализируем формулы (3.68) и (3.70), которые подобны по своей записи, если J' и J'' принимают значения 1, 2, 3,... Разница этих формул состоит только в знаке линейного члена. Если в формуле (3.68) принять J' = m, где m = 1, 2, 3,..., а в равенстве (3.70) положить J'' = –m, то обе формулы становятся одинаковыми и их можно записать в виде
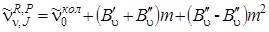 , (3.72)
, (3.72)
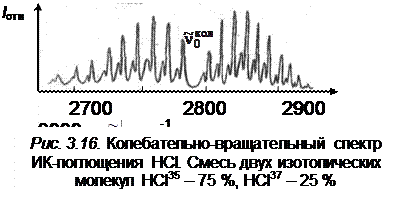
где m = 1, 2, 3,... для R -ветви; m = –1, –2, –3,... для Р -ветви. В соответствии со знаком m часто R -ветвь называют положительной, а Р -ветвь – отрицательной ветвью. Зная расстояния между линиями P - и R -ветвей, можно по формулам (3.73) и (3.70) приблизительно оценить вращательную постоянную, а затем найти равновесное расстояние rе . На рис. 3.16 приведен колебательно-вращательный спектр ИК-поглощения молекулы НСl. Применяя формулу (3.72), нужно всегда помнить, что m > 0 соответствует значению вращательного квантового числа J = J' для верхнего колебательного уровня, a m < 0 – значению квантового числа J = J'' для нижнего,так что симметрия формулы (3.72) не является полной. на рис. 3.16 линии R -ветви расположены справа от колебательной частоты 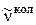 , а Р -ветви – слева. Нулевая линия
, а Р -ветви – слева. Нулевая линия 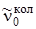 , отсутствующая в колебательно-вращательном спектре, соответствует случаю m = 0. В случае B' u = B'' u формула (3.72) упрощается и принимает вид
, отсутствующая в колебательно-вращательном спектре, соответствует случаю m = 0. В случае B' u = B'' u формула (3.72) упрощается и принимает вид
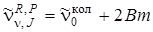 . (3.73
. (3.73
Выражение (3.73) дает равноотстоящие линии, симметрично расположенные относительно чисто колебательной полосы 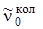 .
.
В случае двухатомных молекул, у которых электронный момент количества движения не равен нулю, наряду с R - и Р -ветвью появляется Q -ветвь (нулевая) (D J = J' – J'' = 0). Для Q -ветви получаем (J = J' = J'')
 |
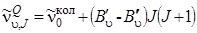 (3.74)
(3.74) Так как разность (B' u – B'' u) мала по сравнению с B' u и B'' u, то линии
Q -ветви расположены в длинноволновую сторону от 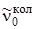 (B' u < B'' u) и при малой дисперсии регистрируемого прибора сливаются в одну узкую полосу.
(B' u < B'' u) и при малой дисперсии регистрируемого прибора сливаются в одну узкую полосу.
3.6.3. Колебательно - вращательные спектры КР
Для спектров КР имеет место правило отбора по вращательному квантовому числу J.
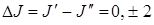 (3.75)
(3.75)
Поэтому в спектре КР наряду с Q -ветвью, состоящей из близкорасположенных линий, будем получать S -ветвь и 0 -ветвь, для которых соответственно 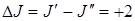 и
и 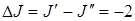 .
.
Выведем общие формулы для положения линий O -, S - и Q -ветвей. В общее выражение для разности вращательных подуровней ветви
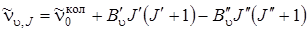 , (3.76)
, (3.76)
подставим вместо J'' выражение через J' из формулы J'' – J' = –2. После этогo получим
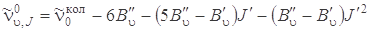 , (3.77) где J' = 0, 1, 2,....
, (3.77) где J' = 0, 1, 2,....
Из анализа формулы (3.77) следует, что линии O -ветви располагаются в низкочастотную сторону от чисто колебательной полосы и будут слегка расходиться вследствие наличия положительного квадратичного члена 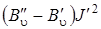 , так как
, так как 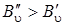 . Аналогичное выражение можно записать для линий S -ветви, если в формуле (3.76) J' заменить на J''. Итак, если J' = J' '+ 2, то
. Аналогичное выражение можно записать для линий S -ветви, если в формуле (3.76) J' заменить на J''. Итак, если J' = J' '+ 2, то
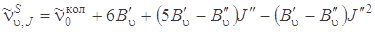 , (1.78)
, (1.78)
где J'' = 0, 1, 2,..., т. е. линии S -ветви будут располагаться в коротковолновую сторону от чисто колебательной полосы и будут слегка сходиться в силу наличия квадратичного члена 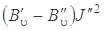 (так как
(так как 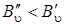 ). Четвертое слагаемое в формуле (3.78) всегда отрицательно, что и приводит к небольшому схождению линий S -ветви при больших J'' (рис. 3.17), где приведены спектры КР молекулы НСl).
). Четвертое слагаемое в формуле (3.78) всегда отрицательно, что и приводит к небольшому схождению линий S -ветви при больших J'' (рис. 3.17), где приведены спектры КР молекулы НСl).
Почему мы в случае О -ветви выражаем J'' через J', а в случае S -ветви, наоборот, J' через J''. Это следует из того, что как в первом случае для О -ветви, так и во втором случае для S -ветви вращательное квантовое число начинается с нуля (в соответствии с D J = ±2). Если бы в формулу (3.77) мы ввели J'', а не J', то J'' изменялось бы от 2 и более, чтобы удовлетворить правилу отбора (J' – J'') = 2. Для линий Q -ветви (J' = J'' = J) будем иметь выражение
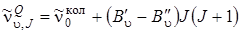 . (3.79)
. (3.79)
Далее, считая что 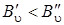 , линии (Q -ветви будут располагаться в длинноволновую сторону от полосы чисто колебательного перехода и могут налагаться на линии О -ветви.
, линии (Q -ветви будут располагаться в длинноволновую сторону от полосы чисто колебательного перехода и могут налагаться на линии О -ветви.
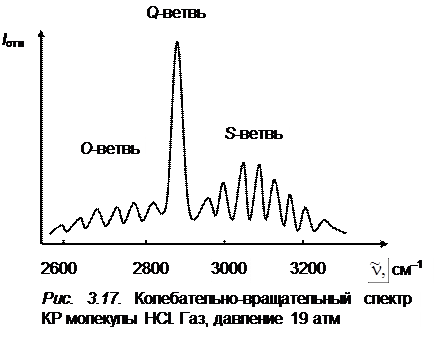
Наиболее интенсивными в спектре КР будут линии Q -ветви (см. рис. 3.17), но так как 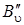 – B'' мало отличается от
– B'' мало отличается от 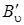 , то линии Q -ветви располагаются очень густо и при малой дисперсии прибора не разрешаются или разрешаются слабо (рис. 3.18, а).
, то линии Q -ветви располагаются очень густо и при малой дисперсии прибора не разрешаются или разрешаются слабо (рис. 3.18, а).
На рис. 3.18, (б) в увеличенном масштабе приводится спектр КР
Q -ветви молекулы водорода. Для вещества, находящегося в твердом или жидком состоянии, как спектры КР, так и спектры ИК-поглощения состоят из одной широкой полосы без следов какой-либо структуры.
Для выяснения общего характера, расположения ветвей вращательных линий в спектрах КР рассмотрим более упрощенный вариант частотных зависимостей ветвей. Пусть 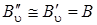 . тогда ветви запишутся следующим образом:
. тогда ветви запишутся следующим образом:
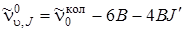 , (3.80)
, (3.80)
где J' = 0, 1, 2,..., т. е. линии О -ветви представляют собой равноотстоящие линии с разностью 4 В. Только нулевая линия (J' = 0) отстоит от чисто колебательной полосы на расстоянии 6 В.

Аналогично для S -ветви получим:
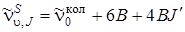 , (3.81)
, (3.81)
где J = 0, 1, 2, 3,..., т. е. линии S -ветви представляют собой равноотстоящие линии, распространяющиеся в область больших частот (расстояние между линиями равно 4 В). Нулевая линия отстоит от центра колебательной полосы на 6 Q в сторону увеличения частот.

