2015-10-16
2015-10-16 1167
1167 Тирозин образуется путем гидроксилирования фенилаланина. Реакция очень сложна, в ней участвуют два печеночных фермента – Е1 и Е2, образующих в совокупности фенилаланилгидроксилазу. Коферментом Е1 является тетрагидробиоптерин (ТГБП Н4- переносчик 2Н+):
Тирозин образуется путем гидроксилирования фенилаланина. Реакция очень сложна, в ней участвуют два печеночных фермента – Е1 и Е2, образующих в совокупности фенилаланилгидроксилазу. Коферментом Е1 является тетрагидробиоптерин (ТГБП Н4- переносчик 2Н+):
1). Фенилаланин + О2 + ТГБП Н4 Тир + Н2О + ДГБП Н2
 2). ДГБП Н2 + НАДФН ТГБП Н4 + НАДФ
2). ДГБП Н2 + НАДФН ТГБП Н4 + НАДФ
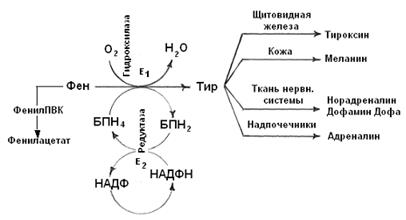
Рис. 8-9. Схема метаболизма фенилаланина и тирозина
При отсутствии Е1 (генетический дефект) или его малой активности развивается состояние называемое фенилкетонурией, которое характеризуется выделением с мочой 1-2 г фенилПВК в сутки. Биохимический механизм этого симптома состоит в том, что фенилаланин дезаминируется, а не превращается в тирозин. Далее возможно декарбоксилирование или восстановление фенилПВК до фенилацетата или фениллактата соответственно. Большие количества этих веществ накапливаются в крови и ткани нервной системы в результате чего у ребенка развивается слабоумие, что сопровождается резко выраженными неврологическими симптомами. При своевременной диагностике (в роддоме) возможна коррекция нарушений применением диеты со сниженным до 20% содержанием фенилаланина в пище.