2017-11-01
2017-11-01 1016
1016Аллель — одна из двух или более альтернативных форм гена, каждая из которых характеризуется уникальной последовательностью нуклеотидов.
Анеуплоидия — отсутствие или избыток одной или нескольких хромосом нормального набора.
Аутосомы — соматические хромосомы.
Гетерозигота - особь с двумя различными типами аллелей в определенном локусе (нормальном и мутантном), находящимся в транс-положении.
Гомозигота — особь с аллелями одинакового типа в определенном локусе (нормальными или мутантными), находящимися в транс-положении.
Гомологичные хромосомы — парные хромосомы диплоидного организма, имеющие одинаковую величину, форму и строение наследственного материала.
Гоносомы — половые хромосомы (X, Y).
Делеция — утрата сегмента ДНК размером от одного нук-леотида до субхромосомного фрагмента, включающего несколько генов хромосомы.
Инверсия - поворот на 180° сегмента ДНК размерами от двух нуклеотидов до субхромосомного фрагмента, включающего несколько генов.
Кариотип — полный набор хромосом диплоидной клетки (кариотип человека в норме 46, XX или 46, XY).
Мозаицизм — присутствие в организме двух или более генетически различных клеточных популяций.
Моносомия — утрата целой хромосомы в хромосомном наборе.
ПЦР - многомиллионное увеличение числа копий определенного участка ДНК in vitro с помощью ферментативного синтеза.
Полисомия — присутствие одной из хромосом более чем в двух экземплярах.
222 3. Бесплодный брак
Полиплоидия — увеличение числа хромосом, кратное гаплоидному набору (3N, 4N и т.д.).
Теломера — концевой участок хромосомы. Специализированная структура, обеспечивающая стабильность линейной молекулы ДНК.
Транслокация — мутация, при которой происходит перемещение гена или участка хромосомы из одного локуса в другой.
Несбалансированная транслокация — транслокация, при которой вместе с участком хромосомы теряется часть уникальных генов, что приводит к патологии.
Реципрокная транслокация — транслокация, при которой происходит обмен участками между двумя хромосомами хромосомного набора.
Робертсоновская транслокация — транслокация акроцент-рических хромосом групп D и G.
Сбалансированная транслокация — транслокация, при которой присутствуют все уникальные гены.
Уникальные гены — гены, представленные единственной копией в гаплоидном наборе хромосом.
Фенотип — совокупность признаков организма, контролируемый определенным генотипом.
FISH — Fluorescent In Situ Hybridization (флуоресцентная гибридизация in situ).
CFTR — cystic fibrosis transmembrane conductase regulator (ген трансмембранного регулятора муковисцидоза).
HLA (Human Leucocyte Antigens)- генетический регион главного комплекса гистосовместимости человека, находящийся на 6-й хромосоме человека и кодирующий антигены системы HLA.
Социально-экономический прогресс в развитых странах, в том числе и в России, повлек за собой принципиальные изменения поведения человека. В последние сто лет все реже встречаются многодетные семьи. В течение репродуктивного периода в жизни женщины только одна или две беременности заканчиваются родами. При этом достаточно часто беременность наступает после 30 лет. В связи с этим возникает тенденция к сохранению часто единственной беременности любой ценой и любыми методами. Необходимо отметить, что в настоящее время эта беременность нередко наступает после применения различных вспомогательных репродуктивных технологий (стимуляция суперовуляции, ЭКО и др.), что создает дополнительные условия для сохранения в популяции
Медико-генетическое консультирование и клинико-генетические методы... 223
предрасположенности к рождению детей с наследственными и врожденными заболеваниями.
Во избежание появления потомства с генетической патологией у этой группы пациентов необходимо проведение профилактических мероприятий, которые обычно реализуются через медико-генетические консультации.
Основные показания для направления супружеских пар на медико-генетическое консультирование: 1) рождение ребенка с наследственными заболеваниями или врожденными пороками развития; 2) наличие у одного из супругов хромосомной перестройки, наследственного заболевания или порока развития; 3) кровнородственный брак; 4) возраст матери старше 35 лет; 5) неблагоприятные воздействия факторов внешней среды в ранние сроки беременности (инфекционные заболевания, особенно вирусной этиологии; массивная лекарственная терапия; ренгенодиагностические процедуры; производственные вредности); 6) наличие самопроизвольных выкидышей и мертворождений неясного генеза, первичной аменореи; бесплодия супругов (после исключения гинекологической патологии); 7) неблагоприятное течение данной беременности (угроза прерывания, многоводие или маловодие, гипотрофия плода, изменения показателей сывороточных маркеров крови матери).
Желательно, чтобы каждая супружеская пара прошла медико-генетическое консультирование до планирования деторождения.
При медико-генетическом консультировании врач-генетик получает сведения о репродуктивной функции самого пациента и его родственников (наследственные заболевания, ранняя детская смертность, врожденные пороки развития, бесплодие, невынашивание беременности и т.д.). На основании полученной информации врач делает заключение о необходимости проведения тех или иных генетических исследований у супружеской пары перед программой ЭКО и ПЭ, ИКСИ. Эти результаты помогут специалисту выяснить причину возникновения у них нарушений в репродуктивной функции и определить риск появления потомства с наследственной или врожденной генетической патологией у супружеской пары.
В данной главе будут рассмотрены показания к проведению тех или иных генетических исследований супружеских пар программы ЭКО и ПЭ, ИКСИ, современные возможности определения ранее не диагностированных генетических нарушений, их влияние на репродуктивную функцию. В за-
Бесплодный брак
ключение будет представлен алгоритм генетического обследования пациентов программы ЭКО и ПЭ, ИКСИ.
Возникновение патологических состояний репродуктивной системы может быть обусловлено хромосомными аномалиями, генными мутациями и наличием наследственной предрасположенности к заболеванию.
Медико-генетическое консультирование показывает, что каждая 8-я супружеская пара с нарушением репродуктивной функции нуждается в цитогенетической диагностике (Loan D., 1987).
В Научном центре акушерства, гинекологии и перинато-логии РАМН было обследовано 669 пациентов (342 женщины и 327 мужчин), включенных в программу ЭКО и ПЭ, ИКСИ. Исследование лимфоцитов крови выявило 32 аномальных ка-риотипа (4,8%). Нарушения кариотипа среди мужчин были обнаружены у 5,5% (или 56% от общего числа лиц с аберрантным кариотипом), среди женщин — 4,1% (или 44% от общего числа лиц с аберрантным кариотипом). У 6 из 18 мужчин с хромосомными нарушениями был обнаружен кариотип, характерный для синдрома Клайнфельтера, который был представлен как полной, так и мозаичной формами. Остальные мужчины имели варианты сбалансированных транслокаций, в которых участвовали аутосомы разных групп.
У 10 из 14 женщин выявлены нарушения в комплексе половых хромосом, характерные для синдрома Шерешевско-го—Тернера, которые были представлены мозаицизмом; один из клонов имел кариотип 45, X. Одна женщина из этой группы имела несоответствие кариотипа 46, XY — женскому фенотипу.
Инверсии хромосом были выявлены и у мужчин, и у женщин. Они были представлены в основном инверсией 9-й хромосомы, которая преобладала в кариотипах женщин. Характер хромосомных нарушений представлен в таблице 1.
Анализ результатов кариологического исследования пациентов программы ЭКО и ПЭ, ИКСИ установил, что самыми частыми нарушениями кариотипа (у 18 из 32) были изменения, связанные с нарушением комплекса половых хромосом. Эти данные совпадают с данными литературы. Большинство авторов обнаружили, что аномалии гоносом составляют до 2/3 всех хромосомных нарушений (Курило Л. и др., 1997).
Наиболее часто встречающимися хромосомными заболеваниями (синдромами), обусловленными нарушением комплекса половых хромосом, являются синдром Шерешевско-
Медико-генетическое консультирование и клинико-генетические методы... 225
--------------------------------------------------------------------------------------------
Таблица 1 Кариологическое исследование пациентов программы ЭКО и ПЭ, ИКСИ
| Число наблюдений с патологией кариотипа (N=32) | Кариотип |
| Мужчины N=18 (56% от общего процента хромосомной патологии) | |
| 47.XXY | |
| 46, XY/47, XXY (3%/97%) | |
| 46, XY/47, XXY (96%/4%) | |
| 47, XYY | |
| 46, XY/47, XYY (96%/4%) | |
| 46, XY/46, XX (94%/6%) | |
| 8 наблюд. (44,4%) | |
| 45, XY, t(13/14)(ql0ql0) | |
| 46,XY,t(3/5)(qterql5) | |
| 46, XY, t(7/16)(q21q22) | |
| 46, XY, t (8/15)(ql3qter) | |
| 7 наблюд. (38,9%) | |
| 46, XY, inv 9 | |
| 46, XY, inv 7 | |
| 2 наблюд. (11,1%) | |
| 1 (5,6%) | 46, XY/47, XY+mar (67%/33%) |
| Женщины 14 (44% от общего процента хромосомной патологии) | |
| 45, X/46, XX (4%/96%) | |
| 46, XX/47, XXX (98%/2%) | |
| 45, X/46, XX/47, XXX (6%/91%/3%) | |
| 45, X/46. XX/47, XXX (6%/92%/2%) | |
| 45, X/46, XX/47, XXX/48, XXXX | |
| (4%/95%/0,7%/0/3%) | |
| 46, Xdel (X)(p21.3) | |
| 45, X/46, Xi (Xq) (20%/80%) | |
| 45, X/46, Xi (Xq)/47, XXi (Xq) (94%/3%/3%) | |
| 46, XX/46, XXq- (96%/4%) | |
| 46, XY | |
| 10 наблюд. (71,4%) | |
| 2(14,4%) | 46, XX, inv 9 |
| 1 (7,1%) | 46, XX, inv 8(pllql3) |
| 1 (7,1%) | 45, XX, t(13/14)(ql0ql0) |
226 3. Бесплодный брак
го—Тернера у женщин, синдром Клайнфельтера и полисомия Y-хромосомы у мужчин.
Синдром Шерешевского—Тернера среди новорожденных девочек встречается с частотой 1:5000. При моносомии Х-хромосомы развивается типичная форма дисгенезии гонад.
Примерно у половины пациентов с синдромом Шерешевского—Тернера обнаруживается хромосомный мозаицизм (45, Х/46, XX) в результате нерасхождения Х-хромосом на ранних стадиях развития эмбриона. В зависимости от соотношения клонов ХХ/Х фенотип больных может проявляться от типичной картины заболевания до нормального фенотипа.
Причиной синдрома Шерешевского—Тернера могут быть структурные нарушения одной из Х-хромосом, которая имеет вид кольцевой хромосомы, изохромосомы, делеции короткого плеча Х-хромосомы или др. У части больных с синдромом Шерешевского—Тернера обнаруживается в кариотипе присутствие Y-хромосомы или кольцевые и маркерные ее фрагменты. Г.Р.Осиповой (1997) было показано, что у 15,4% больных с синдромом Шерешевского—Тернера (кариотип 45, X) в лимфоцитах крови был обнаружен ген SRY, который свидетельствовал о наличии тканевого мозаицизма и присутствии Y-хромосомы. Идентификация этих хромосомных фрагментов имеет принципиальное значение, так как известно, что наличие Y-хромосомы в кариотипе пациенток с синдромом Шерешевского— Тернера часто сопровождается мали-гнизацией дисгенетичных гонад и требует высокой онкологической настороженности. Опухоли дисгенетичных гонад у таких пациентов до 16 лет выявляются в 10,2% случаев; во второй и третьей декадах жизни процент этот возрастает до 30-35% (Розовский И.С. и др., 1977).
Учитывая эти обстоятельства, идентификация Y-хромосомы и ее хромосомных фрагментов чрезвычайно важна, однако это не всегда возможно при использовании классического цитогенетического метода. В этом случае применяют молеку-лярно-цитогенетический FISH-метод, с помощью которого можно идентифицировать хромосомы и определять их количество. Преимущество этого метода заключается в том, что он позволяет не только на метафазных, но и на интерфазных клетках проводить исследование. Кроме того, с помощью этого метода можно проанализировать большое количество клеток, что ограничено при классическом цитогенетическом анализе. Это имеет принципиальное значение при установлении небольшого процента клона клеток с хромосомными нарушениями.
Медико-генетическое консультирование и клинико-генетические методы... 227
Примером практического использования FISH-метода в лаборатории клинической эмбриологии НЦ АГиП РАМН было уточнение принадлежности маркерной и кольцевой хромосом у больных с синдромом Шерешевского—Тернера, что имеет принципиальное значение для выбора метода лечения. В обоих случаях было установлено, что эти фрагменты являются частью Х-хромосомы. Кроме того, этот метод позволил уточнить хромосомный диагноз и определить соотношение клонов у пациентки со сложным хромосомным мозаи-цизмом, который был представлен 46, ХХ/45, Х/47, ХХХ/48, ХХХХ (95% /4% /0,7%/ 0,3% соответственно).
Вышесказанное свидетельствует о важности сочетания методов кариологического исследования и гибридизации in situ для уточнения хромосомного диагноза, особенно у пациентов программы ЭКО и ПЭ, ИКСИ.
В большинстве случаев полная или частичная потеря одной Х-хромосомы ведет к абсолютному бесплодию. Однако в 1960 г. (Banner F.) появилось первое сообщение о фертиль-ности пациентки с синдромом Шерешевского—Тернера и с кариотипом 45, X. С того времени в печати накопилось достаточно много работ, в которых сообщается о наступлении беременностей у женщин с этим синдромом. В частности, F.Din Kelmann и соавт. наблюдали 28 пациенток с синдромом Шерешевского—Тернера, у которых наступила беременность. У 6 из них кариотип был 45, X, у остальных — мозаицизм (45, Х/46, XX; 45, Х/47, XXX; 45, Х/46, ХХ/47, XXX). Обращают на себя внимание высокая частота пороков развития в потомстве и неблагоприятный исход беременностей у женщин с этим синдромом. Более подробно исходы беременностей этих пациенток представлены в таблице 2.
Существует предположение, что наличие репродуктивной функции у пациенток с кариотипом 45, X связано с нераспознанным мозаицизмом, присутствием клона клеток с нормальным кариотипом. Это подтверждают исследования, проведенные A.Novac с соавт. (1995), которые у одной и той же больной обнаружили разные кариотипы в лимфоцитах, кожных фибро-бластах, а также в левой и правой гонадах. Кариотип, определенный в лимфоцитах периферической крови, не в каждом случае определяет ситуацию в гонадах. Приведенные выше данные свидетельствуют, что у женщин с хромосомными мутациями имеется высокий риск созревания яйцеклеток с анеуплоидией.
Частота синдрома Клайнфельтера в популяции — 0,5—2 на 1000 новорожденных мальчиков (полные и мозаичные формы), а полисомии Y-хромосомы — 1 на 1000 (Айала Ф., 1988).
Бесплодный брак
Таблица 2 Исходы беременностей у пациенток с синдромом Шерешевского-Тернера
| Исход беременности | Количество наблюдений |
| Спонтанные аборты | |
| Медицинский аборт | |
| Мертворождение (у одного из плодов обнаружены пороки развития) | |
| Рождение живых детей: | |
| здоровые дети | |
| пороки развития | |
| изолированные пороки | |
| мозга и сердца | |
| синдром Шерешевского—Тернера | |
| синдром Дауна | |
| Состояние детей неизвестно |
Как и при синдроме Шерешевского—Тернера, при синдроме Клайнфельтера и синдроме полисомии Y-хромосомы часто встречается клеточный мозаицизм. Предполагается, что у мужчин с этими синдромами клетки терминального эпителия способны к мейозу и из них могут формироваться сперматозоиды как с нормальным гаплоидным хромосомным набором,так и с анеуплоидией.
Помимо аномалий половых хромосом многие авторы отмечают у пациентов программы ЭКО и ПЭ, ИКСИ повышенный процент сбалансированных хромосомных аберраций аутосом по сравнению с общей популяцией. В общей популяции, по данным P.Jacobs (1974), сбалансированные транслокации встречаются с частотой 0,1%. Среди пациентов программы ИКСИ их частота достигает 6,2% у мужчин и 9,8% у женщин (Van der Ven et al., 1998).
Робертсоновские транслокации составляют большинство транслокаций у бесплодных мужчин, причем наиболее часто в перестройку вовлечены хромосомы 13 и 14. Частота этих аномалий значительно повышена по сравнению с общепопу-ляционным уровнем.
У женщин — носителей сбалансированных хромосомных транслокаций часто наблюдаются особенности менструальной и репродуктивной функций (см. табл. 3 и 4).
Эта группа женщин составляет значительную часть пациентов, нуждающихся в лечении с помощью методов вспомогательной репродуктивной технологии.
Медико-генетическое консультирование и клинико-генетические методы... 229
Таблица 3 Особенности менструальной функции у женщин со сбалансированными хромосомными перестройками (СХП)
| Особенности менструальной | Женщины с СХП | Женщины контроль- |
| функции | п=55 | ной группы п=33 |
| Поздние менархе | 12(21,8%) | 7(21,2%) |
| Менструации установились | ||
| ч/з 3 года | 7(13%) | - |
| Нерегулярный менструаль- | ||
| ный цикл | 6(11%) | 1(3%) |
| Болезненные менструации | 6(11%) | 2(6%) |
| Обильные менструации | 10(20%) | 3(9%) |
Таблица 4 Исходы беременностей у женщин со сбалансированными хромосомными перестройками (СХП)
| Исход беременности | Число наблюдений |
| Самопроизвольное прерывание и неразвивающаяся беременность | 71 (44,9%) |
| Рождение ребенка с пороками развития | 63 (39,9%) |
| Антенатальная гибель плода и смерть ребенка в раннем детском возрасте неясного генеза | 14 (8,9%) |
| Рождение здорового ребенка | 10 (6,3%) |
С введением в практику метода преимплантационной генетической диагностики (ПГД) появилась возможность определения хромосомных нарушений у эмбрионов, родители которых имеют хромосомные мутации и включены в программу ЭКО и ПЭ, ИКСИ. О важности этого метода свидетельствуют случаи рождения здоровых детей после профилактической ПГД у супружеских пар с хромосомными нарушениями в кариотипе.
ПГД проводится в период раннего эмбриогенеза in vitro. В случае, когда у родителей в кариотипе имеется хромосомная транслокация, эта диагностика позволяет исключить наличие патологии, но не может ответить на вопрос, имеется ли в кариотипе эмбриона сбалансированная транслокация, как у родителей, или она отсутствует. Только последующая пренатальная диагностика может это установить.
Перенос эмбрионов со сбалансированным кариотипом исключает рождение детей с хромосомной патологией, спо-
230 3. Бесплодный брак
собствует нормальному развитию беременности и своевременным родам в программе ЭКО и ПЭ, ИКСИ.
До сих пор остается открытым вопрос о влиянии хромосомных вариантов в кариотипе на репродуктивную функцию. К хромосомным вариантам относят увеличенный околоцент-ромерный гетерохроматин отдельных хромосом lqh+; 3qh+; 9qh+; 16qh+, Yqh+, а также увеличенные спутники хромосом групп DuG (13, 14, 15,21,22).
Гетерохроматические (ГХ) районы являются необходимым элементом, эволюционно закрепленным в структуре всех эукариотических организмов — растений, животных и человека. В геноме человека они составляют 16%. До настоящего времени биологическая роль ГХ остается во многом не выясненной. В то же время установлено, что особенности его строения могут оказывать существенное влияние на функционирование хромосом и уникальных генов, расположенных рядом, что часто сопровождается значительными последствиями для организма.
Имеются наблюдения, что увеличение размеров ГХ могут сочетаться с невынашиванием беременности, дефектами развития, а также возникновением хромосомных заболеваний.
J.Nielsen и соавт. (1974), исследуя частоту увеличения гете-рохроматина 9-й хромосомы (9 qh+), установили, что в популяции она равняется 0,3%; у родственников в семьях, где рождались дети с врожденными пороками развития и хромосомными заболеваниями (синдромом Дауна, синдромом Шере-шевского—Тернера и др.), частота этого варианта хромосомы увеличивалась почти в 12 раз (3,5%). S.Holbek и соавт. (1974) установили сочетание аномалий развития и нарушение репродуктивной функции с выраженными изменениями размеров гетерохроматических районов хромосом.
Т.Г.Цветкова (1980) изучала вариабельность структурного гетерохроматина в группе супружеских пар с отягощенным акушерским анамнезом (повторные спонтанные аборты, мер-творождения, рождение детей с пороками развития) и выявила у них увеличение частоты хромосомных вариантов по сравнению с контрольной группой. Кроме того, было отмечено, что хромосомные варианты чаще определялись у женщин, чем у мужчин в этой группе.
Такие же результаты получила Н.А.Каретникова (1981) при исследовании кариотипа супружеских пар с привычным невынашиванием и у пар, в анамнезе которых были дети с врожденными пороками развития. При сопоставлении особенностей кариотипа, с увеличенными вариантами структур-
Медико-генетическое консультирование и клинико-генетические методы... 231
ного гетерохроматина (1, 9, 16 и акроцентрических хромосом), с результатами дерматоглифических исследований, характером родословных и с клинической характеристикой отмечено, что с увеличением в кариотипах родителей содержания структурного гетерохроматина возрастает количество и частота патологических признаков в дерматоглифике, частота отягощенных родословных, тяжесть нарушений репродуктивной функции.
И.В.Бутомо и соавт. (1981) проводили изучение гетерохроматических районов 1,9, 16 и Y-хромосом у детей с болезнью Дауна и отметили у них высокий процент наличия увеличенного гетерохроматина в 9-й хромосоме по сравнению с контрольной группой (16,43% против 9,3% в контроле).
В литературе накоплен обширный материал относительно повышенной частоты вариантов гетерохроматического района хромосомы 9 у детей с множественными пороками, с олигофренией и их родителей.
И.А. Демидова и соавт. (1990) сообщают, что хромосомные варианты довольно часто (до 20%) встречаются у людей при недифференцированных формах умственной отсталости.
Известно, что среди супружеских пар с бесплодием и невынашиванием беременности эти варианты встречаются чаще, чем в популяции. О.Б.Барцева и соавт. (1998) при цитогенети-ческом обследовании супружеских пар, в анамнезе которых имелось несколько неудач в программе ЭКО и ПЭ, ИКСИ, обнаружила высокий процент хромосомных вариантов.
Однако не все исследователи придерживаются этой точки зрения.
Учитывая разноречивость данных, супружеские пары, включенные в программу ЭКО и ПЭ, ИКСИ и имеющие в ка-риотипе хромосомные варианты, требуют более тщательного наблюдения во время беременности. Им рекомендуется более частое проведение УЗИ, исследование фетальных сывороточных маркеров по крови матери; при наличии отклонений в показателях рекомендуется проведение пренатальной диагностики.
Учитывая высокий процент хромосомных аномалий среди супружеских пар с бесплодием, рекомендуется, чтобы всем пациентам программы ЭКО и ПЭ, ИКСИ было проведено цитогенетическое исследование кариотипа по лимфоцитам периферической крови. В случаях подозрения на минимальный скрытый клеточный мозаицизм рекомендуется проведение FISH-анализа по лимфоцитам периферической крови для уточнения диагноза.
232 3. Бесплодный брак
Один из путей успешного решения проблем профилактики заболевания связан с выявлением предрасположенности индивидуума к ним. По данным ряда авторов, антигены системы HLA могут быть успешно использованы для этой цели. В настоящее время накоплен большой фактический материал о возможности использовать их в качестве иммунологических маркеров в генетике, судебной медицине, акушерстве, гематологии, трансфузиологии, трансплантологии и других областях. На основании этого появилось новое направление в медицине «HLA и болезни».
Система HLA (Human Leucocyte Antigens) является частью главного комплекса гистосовместимости МНС (Major Histocompatibility Complex) и располагается на коротком плече 6-й хромосомы.
Согласно современным представлениям, гены системы HLA относят к трем классам. К I классу относят локусы HLA -А, -В, -С, ко II классу - локусы HLA-D/DR, -DQ, -DP, -DN и DO. Регион генов III класса содержит аллели компонентов комплемента С2 и С4, пропердинового фактора (Bf), фактора некроза опухоли (TNF), изоферментов и др., регулирующих иммунные процессы в организме.
Каждый локус представлен серией аллельных генов с разной степенью частоты распределения в человеческих популяциях. Гены HLA имеют кодоминантное выражение, благодаря чему антигены HLA каждого локуса, полученного от обоих родителей, могут быть обнаружены у обследуемого.
В 1980 г. на основании популяционных исследований было сделано 3 главных вывода:
1) антигены гистосовместимости являются общими для всех популяций, населяющих Землю;
2) имеются определенные количественные различия в распределении антигенов гистосовместимости, когда одни и те же антигены в одних расах имеют высокую фенотипиче-скую частоту, а в других представлены лишь в следовых количествах;
3) антигены, представленные в различных расах, в отдельных этнических группах имеют разную фенотипическую частоту, которая характерна для каждой этнической группы этих рас.
Таким образом, антигены HLA обладают определенной специфичностью, что надо учитывать при использовании их в качестве генетических маркеров.
Продукты генов HLA представляют собой антигены на поверхности клеток, органов и тканей, в том числе и лимфоци-
Медико-генетическое консультирование и клинико-генетические методы... 233
тах, выполняющие роль рецепторов, которые участвуют в регуляции иммунного ответа, помогая отличать собственные и чужеродные структуры. Генетические структуры (гены-антигены) могут быть определены серологическими методами с помощью HLA-сывороток или генетическими (ДНК).
В 1977 г. B.Duppont и соавт., изучая семьи с врожденной дисфункцией коры надпочечников, обусловленной только недостаточностью 21-гидроксилазы (ВДКН-21), обнаружили, что все пораженные в семье имеют идентичный набор антигенов HLA. На основании этого был установлен факт сцепления генов HLA с мутантными аллелями гена СУР21В, кодирующего фермент 21-гидроксилазы (21-ОН). Недостаточность 21-ОН является причиной самого распространенного варианта ВДКН, который составляет 95% от всех форм ВДКН, относится к группе аутосомно-рецессивных наследственных заболеваний. Особенностью данной формы ВДКН является клинический полиморфизм, в основе которого лежит генетический гетероморфизм. Классические формы заболевания характеризуются тяжелым течением, сопровождаются высокой смертностью новорожденных и врожденными пороками наружных половых органов у девочек. Мягкие формы могут проявляться в разном возрасте, что связано с активностью 21-ОН, которая зависит от особенности мутации гена СУР21В. Эти формы у женщин сопровождаются ранним ад-ренархе, гирсутизмом, нарушениями менструального цикла, ановуляцией, развитием поликистоза яичников (ПКЯ), бесплодием. Частота мягких форм ВДКН-21 составляет 1:100. Однако в разных этнических группах частота мягких форм значительно отличается от средне популяционных (табл. 5) (NewM. et al., 1984).
Э.Р.Дуринян (1997) обнаружила у 61% женщин с ПКЯ но-сительство мутантных аллелей гена СУР21В.
Открытие B.Duppont позволило использовать антигены системы HLA как фенотипические маркеры для диагностики носительства мутантных аллелей гена СУР21В при проведении генеалогических исследований. Широкие популяцион-ные исследования семей, имеющих больных с ВДКН, обнаружили фенотипы HLA, ассоциирующиеся с мутациями гена, которые являлись специфичными для разных этнических групп (табл. 6).
Благодаря полученным данным появилась возможность проводить проспективный поиск мутаций с нарушениями репродукции и гиперандрогенией, формировать группу риска для исследования функции коры надпочечников с помощью
234 3. Бесплодный брак
Таблица 5 Частота встречаемости ВДКН в различных этнических группах
| Классические формы среди новорожденных | Мягкие формы среди гетерозиготных носителей | ||
| США Мариленд | 1:67 000 | Евреи Ашкенази | 1:27 |
| США Аляска | 1:700 | Испанцы | 1:40 |
| США эскимосы | 1:245 | Итальянцы | 1:300 |
| Англия (Бирмингем) | 1:7255 | Славяне | |
| Германия | 1:9831 | ||
| Австрия (Тироль) | 1:8991 | ||
| Швейцария | 1:15 472 |
Таблица 6 Фенотипы HLA, ассоциирующиеся с ВДКН-21, в разных этнических группах
| Этническая группа | Фенотип HLA | Источник |
| Финны | A3 В40 DR1 | PartenenJ. et al, 1989 |
| Итальянцы | В51 | Belvedere M.et al.. 1984 |
| Венесуэльцы | В39, Bw62 | Layrisse Z. et al., 1987 |
| Французы | А10В35, АЗВ15 | Coullin P. et al., 1980 |
| Поляки | A3 В40, A3 В35 | Cruz-Marin F. et al., 1981 |
| Японцы | All B15DR4 | Dzevris I., Ginalska-Malinowska M., 1990 |
| Немцы | Bw47, B35 | Harada Fumiki. 1987 |
| Русские | В14 DQA1 0101/0102 B35, A3 B47 | Bochm B.O., 1987 Dzenis I., Evgrafov O.V., 1995 |
пробы с АКЛТ или молекулярно-генетического исследования. Это крайне важно из-за высокой вероятности у них рождения ребенка с ВДКН.
Антигены системы HLA как генетические маркеры приобрели важное значение для выявления предрасположенности к возникновению ряда заболеваний, связанных с мужским бесплодием.
D.Aleksovski и соавт. (1988) при исследовании мужчин югославов с азооспермией и олигозооспермиеи неизвестной этиологии выявили повышенную ассоциацию антигенов системы HLA с нарушением сперматогенеза. Наиболее частыми антигенами локуса А были А26 и А28, локус В был представлен только антигеном В18.
В.С.Михайличенко и соавторы (1992) исследовали у мужчин — жителей Санкт-Петербурга ассоциации антигенов HLA
Медико-генетическое консультирование и клинико-генетические методы... 235
системы с бесплодием, обусловленной левосторонним вари-коцеле. Обследование 97 бесплодных мужчин с различной степенью олигозооспермии (37 — с I степенью, 36 — со II степенью и 24 — с III степенью) выявило повышение частоты встречаемости антигенов по локусу А — A3, All, AW19, а по локусу В - В13, В16, В22, В27, В35, а также снижение частоты В5. Наиболее выраженной генетической связью с бесплодием при варикоцеле обладают антигены A3, В13, В22. Они также показали, что наиболее сильную гаметную ассоциацию у этих больных дают гаплотипы АЗ-В22, Al 1-B22, AW19-B22.
Повышенная ассоциация антигенов системы HLA с идио-патической азооспермией была найдена также H.Miura и со-авт. (1998) у 65 японских мужчин. Частота антигенов HLA АЗЗ, В13 и В44 была значительно выше по сравнению с контрольной группой. Авторы предполагают, что антигены I класса системы HLA являются важными генетическими маркерами предрасположенности к возникновению идиопатической азооспермии.
В последние годы все большее внимание уделяется поиску причин привычного невынашивания беременности и бесплодия неясного генеза.
Исследование Т.В.Карповой (1998), изучавшей женщин, страдающих невынашиванием беременности и бесплодием неясного генеза, принадлежавших к русской популяции, обнаружила особенности фенотипов HLA при данной патологии. Было также выявлено увеличение частоты антигенов HLA-A19, В8, В13, В15, В35, DR5, DR7 у этой группы - 19,0; 9,5; 19,0; 17,5; 22,2; 69,6 и 39,1% соответственно по сравнению со здоровыми женщинами, частота которых составляла 6,3; 3,8; 10,3; 16,7; 29,9 и 22,7% соответственно. Более подробно данные представлены в таблице 7.
Кроме того, следует учитывать HLA-фенотипы женщины и ее мужа.
В настоящее время одну из причин невынашивания беременности связывают с наличием идентичности HLA супругов не менее чем по двум антигенам. Наиболее неблагоприятная картина складывается при наличии 3 или 4 идентичных антигенов в фенотипах супругов. По данным Т.В.Карповой, ведение беременности у этих пар требует сохраняющей терапии. При наличии двух идентичных антигенов в фенотипах супругов частота неблагоприятного исхода беременности без лечения увеличивается с каждым последующим выкидышем и составляет после первого выкидыша 40%, после второго — 83% и после третьего — 100%.
236 3. Бесплодный брак
Таблица 7 Встречаемость HLA у женщин при привычном невынашивании беременности и бесплодии
| HLA | Патология, п=82 (%) | Физиологическое течение беременности, п=79 (%) | |
| Привычное невынашивание | Бесплодие | ||
| А19 | 19,0* | 16,7 | 6,3 |
| A3 | 20,0 | 44,4 | 20,3 |
| В8 | 9,5 | 11,1 | 3,8 |
| В13 | 16,7 | 10,3 | |
| В15 | 17,5 | 31,6* | 10,3 |
| В35 | 22,2 | 31,6 | 16,7 |
| DR5 | 69,6** | 29,9 | |
| DR7 | 39,1 | 62,5 | 22,7 |
| A3, В35 | 11,1 | 16,6 | 1,3 |
| А1,В8 | 6,3 | 11,1 | 1,3 |
| А19, В15 | 6,3 | 11,1 | 1,3 |
| А2, В15 | 11,1 | 22,2 | 3,8 |
* Различия статистически достоверны, р<0,05. ** Различия статистически достоверны, р<0,01.
Таким образом, всем пациентам программы ЭКО и ПЭ, ИКСИ необходимо проводить HLA-типирование и результаты учитывать при лечении супружеских пар данным методом. Это позволит сократить процент выкидышей и предотвратить рождение детей с наследственной патологией, тем самым повышая эффективность данного метода лечения.
Причиной полного или частичного угнетения процесса сперматогенеза может явиться деления Y-хромосомы. Ген, контролирующий гаметогенез у мужчин, расположен в эухро-матиновой части длинного плеча Y-хромосомы — фактор азооспермии (AZF).
В AZF-участке Y-хромосомы определяются три неперекрывающихся локуса: AZFa, AZFb, AZFc, делеции в которых сопровождаются нарушениями сперматогенеза различной степени тяжести. Позднее M.Kent-Ferst с соавт. (1999) идентифицировали еще один регион - AZFd, расположенный между AZFb
Медико-генетическое консультирование и клинико-генетические методы... 237
и AZFc участками. В дальнейшем в каждом локусе были обнаружены гены, принимающие участие в гаметогенезе.
Делении локуса AZFa встречаются довольно редко и наиболее часто связаны с азооспермией, проявляющейся синдромом «только клеток Сертоли типа 1» (СКС1) и реже с тяжелой олигозооспермией. СКС1 характеризуется полным отсутствием сперматогенного эпителия внутри семенных канальцев, возникшим в результате первичного дефекта в период раннего сперматогенеза.
Делеции, охватывающие весь регион AZFb, связаны с азооспермией, возникшей в результате прекращения мейоза и созревания сперматозоидов на стадии сперматоцитов. В таких случаях пункция яичка, как правило, не позволяет получить зрелые сперматозоиды. Делеция части региона AZFb приводит к возникновению олигозооспермии.
Делеции локуса AZFc являются наиболее распространенными и приводят к различным нарушениям сперматогенеза: от слабой олигозооспермии до крипто- и азооспермии. При этом у большинства пациентов с делецией локуса AZFc и азооспермией при пункции яичка или эпидидимуса могут быть получены и успешно использованы в циклах ИКСИ те-стикулярные сперматозоиды. Делеции, включающие весь AZFc регион и выходящие за его границы (AZFb+c и AZFa+b+c), по мнению многих авторов приводят к полному отсутствию тестикулярных сперматозоидов.
Делеции локуса AZFd могут иметь широкий диапазон нарушений сперматогенеза от тяжелой тератозооспермии до олигозооспермии.
Эти данные свидетельствуют о том, что размер и характер микроделеций могут иметь прогностическое значение в плане возможности получения гамет для проведения ИКСИ. Обнаружение микроделеций у пациентов позволяет избежать бессмысленного гормонального и хирургического лечения. Многие авторы рекомендуют проводить исследование AZF-локусов у всех бесплодных мужчин при концентрации сперматозоидов менее 5 млн/мл и при азооспермии.
В настоящее время нет данных о негативном влиянии AZF-микроделеций на частоту оплодотворения, дробления и имплантации в результате применения метода ИКСИ. При этом возникает проблема наследования сыновьями данной патологии. В настоящее время известно о многих случаях передачи делеции от отцов сыновьям при лечении бесплодия методом ЭКО и ПЭ, ИКСИ. Однако большинство родителей сознательно пошли на риск, зная о нем заранее.
238 3. Бесплодный брак
Учитывая вышесказанное, супружеским парам (если у отцов обнаружены делении AZF-локусов) необходимо проводить преимплантационную диагностику с целью переноса эмбрионов женского пола.
Мужчин с обструктивной азооспермией при двустороннем и одностороннем отсутствии семявыносящих протоков необходимо тестировать на наличие мутации в гене муковисцидо-за. Мутации гена трансмембранного регулятора муковисци-доза (cystic fibrosis transmembrane conductase regulator, CFTR) сопровождаются азооспермией и аномалиями семявыносящих протоков и эпидидимиса. Врожденное двустороннее отсутствие семявыносящих протоков среди бесплодных мужчин встречается с частотой 1—2%. Муковисцидоз (MB) относится к аутосомно-рецессивным заболеваниям, обладающим высоким клиническим полиморфизмом, в основе которого лежит генетический гетероморфизм. Носительство MB достаточно широко распространено с частотой 1/25 жителей в североевропейской популяции.
Ген MB локализован на длинном плече 7-й хромосомы (7q31), содержит 27 экзонов и его длина составляет 250 кб. Описано более 900 мутаций в гене MB. Разные мутации в гене CFTR могут приводить к различной степени инактивации функции хлорного канала, что, в свою очередь, напрямую коррелирует со степенью тяжести течения заболевания.
Мутация deltaF508 встречается примерно в 70% случаев всех мутаций в популяции и представляет собой делецию трех пар оснований в экзоне 10.
Мягкие формы MB возникают в результате мутации Rl 17H, в гомозиготном состоянии или в компаунде с другими аллелями, в том числе с deltaF508. Они часто обнаруживаются у пациентов с врожденной закупоркой семявыводящих канальцев (vas deferens). При этом клиника муковисцидоза у таких пациентов, как правило, отсутствует или стерта.
P.Jezeguel и соавт. (2000), исследуя 47 пациентов с патологией семявыводящих протоков на наличие мутаций в гене CFTR, отметили, что наиболее частыми были мутации deltaF508 (44,7%) и аллель 5Т (36,2%). Аллель 5Т вызывает снижение нормального уровня CFTR мРНК. Частота этого аллеля в общей популяции составляет около 5%. Авторы пришли к выводу, что комбинация в гене CFTR 5T аллеля и какой-либо мутации, характерной для муковисцидоза, в парном аллеле является наиболее частой причиной врожденного двустороннего отсутствия семявыводящих протоков. Аллель 5Т, по мнению некоторых авторов, может являться причиной на-
 Медико-генетическое консультирование и клинико-генетические методы... 239
Медико-генетическое консультирование и клинико-генетические методы... 239
рушения сперматогенеза различной степени тяжести и идио-патического бесплодия у женщин, особенно в тех случаях, когда аллель находится в гомозиготном состоянии.
В связи с потенциально фатальной природой данного заболевания для потомства исследование на мутации гена MB должно стать правилом в случае подозрения на врожденные поражения семявыносящих протоков или эпидидимиса у отца.
Введение в медицинскую практику процедуры ИКСИ позволило мужчинам с отклонениями в спермограмме иметь своих биологических детей. Однако одним из наиболее обсуждаемых вопросов, связанных с данной методикой, остается безопасность данного метода для потомства. У мужчин, включенных в программу ИКСИ, самыми распространенными нарушениями сперматогенеза являются олиго-, астено-, терато-, зооспермии, которые могут встречаться по отдельности или в сочетанной форме — олигоастенотератозооспер-мии (ОАТ). Различные отклонения в морфологии сперматозоидов считаются противопоказанием к проведению ИКСИ, так как предполагается, что сперматозоиды при ОАТ подвержены более частым нарушениям мейоза. Поэтому в последнее время в мире уделяется большое внимание исследованию уровня хромосомных аберраций в половых клетках пациентов с нарушением репродуктивной функции. Представления о характере хромосомных аномалий в мужских половых клетках необходимы для понимания механизмов их происхождения и наследования. Полученные данные служат основой для разработки методов профилактики хромосомной патологии.
Исследования генетической патологии сперматозоидов проводят с помощью FISH-метода с применением специфических зондов на различные хромосомы. Многие авторы применяют трехцветный FISH-анализ. Чаще используют зонды на Х-, Y-хромосомы с добавлением зонда на соматическую хромосому. Добавление пробы на соматическую хромосому необходимо дня дифференцирования диплоидного набора клетки от дисомии половых хромосом.
Исследования сперматозоидов у мужчин с анеуплоидией в кариотипе по лимфоцитам крови (полные и мозаичные формы синдрома Клайнфельтера и синдрома полисомии Y) с применением трехцветного FISH анализа (зонд на X, Y и 18 хромосомы) на анеуплоидии, по данным разных авторов, показало у них высокий процент сперматозоидов с патологическим кариотипом по сравнению с нормой — 2,31—3,9% против 0,43-1,46%. Эти данные о повышенном проценте анеуп-
Бесплодный брак
лоидий в сперматозоидах пациентов с синдромом Клайн-фельтера и синдрома полисомии Y совпадают с данными других авторов, которые также отмечают высокий процент генетической патологии в половых клетках этих пациентов. Учитывая высокий процент сперматозоидов с хромосомными аберрациями у этих больных, выше перечисленные авторы рекомендуют исследовать у них сперматозоиды перед проведением процедуры ИКСИ для установления потенциального риска появления в их потомстве ребенка с генетической патологией.
Изучение хромосом в сперматозоидах, полученных от мужчин с ОАТ, включенных в программу ИКСИ, выявило повышенный процент анеуплоидий по сравнению с контрольной группой. По данным M.Pang (1999), частота дисомий аутосом составила 5,4% у мужчин с ОАТ по сравнению с 0,05-0,2% в контроле; частота диплоидных сперматозоидов у пациентов с ОАТ была 0,4-9,6%; в контрольной группе — 0,04%. По данным L.Colombero и соавт. (1999), эта закономерность также была найдена. Однако процент анеушюидий был несколько ниже, а показатели контроля значительно выше: 2,7% у мужчин с ОАТ против 1,8% в контроле.
По данным нашей лаборатории процент анеуплоидий в сперматозоидах мужчин с ОАТ составил в среднем 0,62%, это значение превышает в 2,5 раза значение контрольной группы (0,25%). В своей работе мы использовали зонды к X-и Y-хромосомам фирмы Vysis. Исследуя сперматозоиды на анеуплоидий только при астенозооспермии нами не было найдено различий в результатах опытной и контрольной групп. Некоторые авторы также отмечают, что при низкой подвижности сперматозоидов не наблюдается повышенный процент сперматозоидов с хромосомной патологией.
Учитывая результаты исследований, все вышеперечисленные авторы предполагают, что использование сперматозоидов пациентов с олигоастенотератозооспермией в программе ИКСИ может привести к увеличению частоты хромосомных патологий у потомков. Уже опубликованы работы, посвященные пренатальной диагностике плодов, полученных с помощью ИКСИ. Исследователи показали, что риск патологии по половым хромосомам повышается до значения 1%, a D. van Opstal и соавт. (1997) доказали, что это обусловлено отцовской патологией. Некоторые авторы полагают что, хромосомные мутации de novo после ИКСИ встречаются чаще, чем в популяции. Они отметили, что риск малых врожденных пороков развития был 2,5 раза выше у мальчиков, чем у девочек.
Медико-генетическое консультирование и клинико-генетические методы... 241
Схема 1 Алгоритм генетического обследования пациентов программы ЭКО и ПЭ, ИКСИ
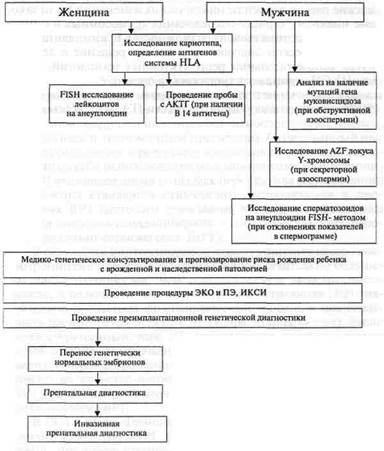
На основании приведенных выше данных многие авторы указывают на необходимость проведения пациентам программы ИКСИ преимплантационной диагностики. Это позволит повысить процент успешной имплантации, уменьшить риск спонтанного аборта и предупредить рождение ребенка с генетической патологией у пациентов программы ЭКО и ПЭ, ИКСИ. Вышеперечисленные авторы также рекомендуют последующее проведение пренатальной диагностики пациентам программы ИКСИ.
Бесплодный брак
Таким образом, большую роль перед введением пациентов в программу ЭКО и ПЭ, ИКСИ играет генетическое консультирование. Очень важно объективно информировать супружеские пары о характере генетических изменений (если таковые имеются), о риске наследования хромосомных и генных нарушений потомством, о возможностях преимплантацион-ной генетической диагностики, так как решение о лечении путем вспомогательных репродуктивных технологий, включая ИКСИ, принимает супружеская пара.
И в заключение предлагаем алгоритм генетического обследования пациентов программы ЭКО и ПЭ, ИКСИ (схема 1).