2014-01-31
2014-01-31 1260
1260Принята международным союзом биохимиков в 1961 году в Москве. По этой системе ферменты делятся на шесть основных классов, согласно общему типу реакций, которые они катализируют. Классы делятся на подклассы и подподклассы, чтобы можно было описать специфичность каждого индивидуального фермента. Каждому ферменту приписывают код или шифр, например: 2.1.3.4., где первая цифра – номер класса, вторая цифра – номер подкласса, третья цифра – номер подподкласса, четвертая – порядковый номер в подподклассе. Например: 1,1,1,1 – алкоголь-НАД-оксидоредуктаза (алкогольдегидрогеназа); 3,5,1,5 – карбамидамидогидролаза (уреаза).
Различают следующие классы:
1) Оксидоредуктаза (катализирует окислительно-восстановительные реакции всех типов). Деление на подклассы: дегидрогеназы, оксигеназы и так далее (то-есть по типу окисления или восстановления):
а) аэробные дегидрогеназы (оксидазы);
б) анаэробные дегидрогеназы (RН2 + Х = ХН2 + R);
в) цитохромы (переносят электроны);
г) пероксидазы (окисляют с помощью перекисей);
д) каталаза (2 Н2О2 = 2 Н2О + О2);
е) гидроксилазы и оксигеназы (катализируют окисление субстрата путем внедрения в субстрат одного или двух атомов кислорода)
2) Трансферазы (катализируют перенос отдельных групп атомов от донорной молекулы к акцепторной). Деление на подклассы происходит в зависимости от характера переносимых групп: аинотрансферазы (NН2), метилтрансферазы (СН3) формилтрансфераза, ацилтрансфераза и так далее.
3) Гидролазы (катализируют разрыв химических связей с присоединением молекул воды по месту разрыва, то-есть реакции гидролиза). Деление на подклассы зависит от характера разрываемой связи: пептидазы (пепсин, трипсин), эстеразы (сульфоэстеразы, фосфоэстеразы), гликозидазы (амилаза, сахараза, лактаза), фосфотазы, амидазы и так далее.
4) Лиазы (катализируют разрыв химической связи без присоединения воды по месту разрыва, при этом в субстратах образуются двойные связи). На подклассы делят от характера разрываемой связи: - C – C -; - C – N -;
- C – O - и т. д. Например альдолаза разрывает – С – С - связь в сахарах. Декарбоксилоза отщепляет карбоксильную группу, дегидротазы (отщипляют воду).
5) Изомеразы (катализируют взаимопревращение различных изомеров). Деление на подклассы зависит от формы: L – D, цис-, транс-, альфа – бета, лактим-лактам (эпимеразы, таутомеразы, рацимазы) и т.д.
6) Лигазы (синтетазы) – катализируют образование нового вещества из двух других веществ. При этом используется молекула АТФ или другого макроэрга для обеспечения энергией. Деление на подкласы осуществляется в зависмости от того какие вещества вступают в реакцию конденсации (например, глутаминсинтетаза).
ПРИРОДА КАТАЛИЗА.
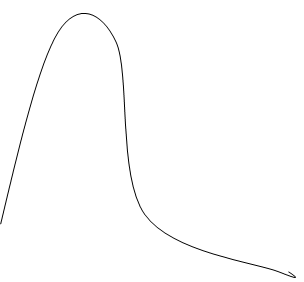
Известно, что результатом любой химической реакции является образование продуктов реакции. Согласно теории «переходного состояния» продукты реакции образуются лишь после того, как реагирующие частицы: 1) – встречаются в определенной пространственной ориентации, и 2) - обладают достаточной энергией, чтобы достичь «переходного состояния» для которого возможны одновременное образование новых и разрыв старых химических связей. Очевидно, что чем легче достигается «переходное состояние», тем выше скорость реакции. Разность между общей энергией исходных реагирующих частиц и энергии возбужденного переходного состояния называется энергией активации, которая характеризует данную химическую реакцию и определяет условия, при которых она происходит.


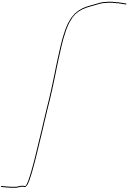 С Переходное состояние (1) - субстрат
С Переходное состояние (1) - субстрат
В (2) - продукт
О (3) – общая энергия
Б (4) – энергия активации
 О
О
Д
Н (4)
А
Я
Э
 Н ____(1)_______________________
Н ____(1)_______________________
Е
Р (3)
Г _________________________________ (2)
 И
И
Я Время протекания реакции
Согласно теории переходного состояния катализатор увеличивает скорость химической реакции, изменяя ее путь так, что новый путь характеризуется более низкой энергией активации. Таким образом вероятность достижения переходного состояния повышается и скорость реакции увеличивается. Хотя энергия активации при действии фермента ниже, энергия общая не меняется.
 Переходное состояние без катализатора
Переходное состояние без катализатора










С
В
О
Б (1)
О
Д
Н Переходное состояние в присутствии катализатора
 А
А
Я (1) – Энергия активации
(2) (2) – Энергия активации 2
Э (3) – Общая энергия - колличество
Н выделяемой энергии
Е
Р Субстрат (3)
Г
И
Я Продукт

Время протекания реакции
Будучи катализатором, ферменты действительно уменьшают энергию активации. Однако, ферментативный катализ характеризуется еще одной особенностью – способностью фермента связывать и ориентировать реагирующие молекулы по отношению друг к другу, так, чтобы образование продуктов реакции происходило максимально эффективно (это одно из положений переходного состояния о котором мы уже говорили). С выполнением этих функций ферментов связан определенный участок на его поверхности, который называют активным центром. Но о нем речь позднее. Хотя детальный механизм действия каждого фермента уникален, все ферменты «работают» сходным образом. Первый подход к изучению действия ферментов разработал Генри (1903г.) и позднее Михаэлис и Ментен (1913г.). Генри, а также Михаэлис и Ментен предложили практически одну и ту же модель, однако Михаэлис и Ментен обосновали свой подход данными тщательно проведенных эксппериментов. Внастоящее время модель Генри-Михаэлиса-Ментена лежит в основе ферментативной кинетики.
МЕХАНИЗМ ДЕЙСТВИЯ ФЕРМЕНТОВ.
На скорость химической реакции влияют различные факторы – концентрация веществ, температура, химическая природа реагирующих веществ. Скорость зависит от наличия активных молекул, поскольку только они могут участвовать в реакциях.
 |
Nакт = Nобщее х е –Е/RT х
№ общее – концентрация вещества
l – постоянная величина основания логорифма общего типа представляют собой мощные катализаторы многих органических реакций, протекающих в водных средах.
Е – энергия активации системы. Этот параметр характеризует природу реагирующих веществ. Чем выше Е, тем меньше скорость реакции, так как становится меньше число активных молекул.
R – универсальная газовая постоянная
Т – температура системы (в градусах Кельвина). Чем выше температура, тем больше скорость реакции, так как увеличивается количество активных молекул.
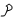 -стерический коэффициент, его величина от 0 до 1, то-есть это число удачных столкновений. Он вводится в формулу для органических соединений.
-стерический коэффициент, его величина от 0 до 1, то-есть это число удачных столкновений. Он вводится в формулу для органических соединений.
 Ферменты уменьшают энергию активации. Например, для реакции 2 Н2О2 2 Н2О + О2 энергия активации составляет 18000 кал / моль. При применении в качестве катализатора платины энергия уменьшается до 11000 кал/моль. Если не работает фермент каталаза, энергия снижается до 1700 кал / моль. Ферменты повышают стерический коэффициент. То-есть фермент одновременно влияет и на энергию активации и на стерический коэффициент, особенно это справедливо для бимолекулярных реакцийю
Ферменты уменьшают энергию активации. Например, для реакции 2 Н2О2 2 Н2О + О2 энергия активации составляет 18000 кал / моль. При применении в качестве катализатора платины энергия уменьшается до 11000 кал/моль. Если не работает фермент каталаза, энергия снижается до 1700 кал / моль. Ферменты повышают стерический коэффициент. То-есть фермент одновременно влияет и на энергию активации и на стерический коэффициент, особенно это справедливо для бимолекулярных реакцийю
Энергия активации сснижается в следствие образования фермент-субстратных комплексов. Эти комплексы не стойкие и распадаются с образованием продукта реакции и фермента в неизменном виде и количестве:

 F + S FS FS* P + F
F + S FS FS* P + F
 В образовании фермент-субстратного комплекса участвуют разные связи (водородная, гидрофобная, электростатического взаимолействия и так далее). Фермент-субстратный комплекс представляет собой реально существующие химические соединения. Некоторые из таких соединений действительно удалось выделить. Фермент-субстратный комплекс образуется в результате связывания фермента с субстратом, то-есть подтверждается основополагающая идея, что при выполнении любой динамической функции белка первым этапом неизменно является связывание. Без связывания белок не может функционировать. И, действительно, связывание, как правило, единственный фактор, определяющий специфичность действия ферментов. Следующим событием в процессе превращения FS F + P является химическая реакция разрыва и образование связи с образованием продукта.
В образовании фермент-субстратного комплекса участвуют разные связи (водородная, гидрофобная, электростатического взаимолействия и так далее). Фермент-субстратный комплекс представляет собой реально существующие химические соединения. Некоторые из таких соединений действительно удалось выделить. Фермент-субстратный комплекс образуется в результате связывания фермента с субстратом, то-есть подтверждается основополагающая идея, что при выполнении любой динамической функции белка первым этапом неизменно является связывание. Без связывания белок не может функционировать. И, действительно, связывание, как правило, единственный фактор, определяющий специфичность действия ферментов. Следующим событием в процессе превращения FS F + P является химическая реакция разрыва и образование связи с образованием продукта.
В ходе образования фермент-субстратного комплекса происходит перераспределение внутримолекулярной энергии за счет смещения электронов, происходит поляризация связей, ослабляющая их (это те связи, которые в процессе катализируемой реакции должны разрушаться). Тем самым снижается энергия активации. Это может быть электрофильная NH3+; Mg++. Или нуклеофильная -ОН, N-- атака, смещения кислотно-щелочного

 равновесия (ОН-,Н+),
равновесия (ОН-,Н+),
NH
устранения стабилизирующей роли воды и так далее. Распад фермент-субстратного комплекса происходит с высокой скоростью.
Размер молекулы субстрата субстрата намного меньше, чем молекула фермента (например, молекулярная масса каталазы равна 250000), поэтому взаимодействие субстрата происходит не со всей молекулой фермента, а с определенным ее участком называемом активным центром. Активный центр может быть один, а может их быть несколько, чаще до четырех. Если фермент – олигомер то активный центр может быть расположен и на одной и на нескольких субъединицах. Участки, удаленные от активного центра представляют собой аллостерические зоны.
 |
|
| R161 |
 R162 R160
R162 R160
 | | S
| | S
R170 R163
R1 R2
| |
R3
|


 По химической структуре активный центр это часть молекулы фермента имеющая сложную пространственную конфигурацию и образованная радикалами входящих в состав фермента аминокислот. Наиболее часто в активный центр входит карбоксильные группы аспарагиновой и глутаминовой кислот, амино-группы аспаргина и лизина, тио-группы цистина и цистеина, гидроксо-группы серина, N-- -группы гистидина. Входят ионы
По химической структуре активный центр это часть молекулы фермента имеющая сложную пространственную конфигурацию и образованная радикалами входящих в состав фермента аминокислот. Наиболее часто в активный центр входит карбоксильные группы аспарагиновой и глутаминовой кислот, амино-группы аспаргина и лизина, тио-группы цистина и цистеина, гидроксо-группы серина, N-- -группы гистидина. Входят ионы
NH
Металлов (цинк, медь, кобальт, калий, натрий, кальций, магний и так далее), входят простетические группы. Все эти радикалы, металлы и простетические группы определенным образом ориентированы в пространстве. Некоторые из них принимают участие в связывание субстрата, другие - в химических превращениях субстрата в продукт. Некоторые группы способны участвовать в обоих процессах. Большинство этих групп занимает удаленное положение в составе полипептидной цепи и пространственно сближены только благодаря многочисленным изгибам, поворотам и скручиванию полипептидной цепи. Очевидно, что аминокислотные остатки, ответственные за связывание и каталитический процесс весьма существенны для активности фермента. Однако не меньшее значение имеют структурные радикалы, благодаря упорядоченному взаимодействию которых образуется и стабилизируется структура молекулы в целом и которые, таким образом, участвуют в формирование активного центра. Свой вклад вносит и ко-фактор. Оптимальная конформация и, следовательно, максимальная активность зависит от выполнения физиологических условий в которых белок сохраняется в интактном состоянии (температура приблизительно 35 – 37градусов по Цельсию, оптимум рН (в среднем 6,5 – 7,5) и так далее). Исключения составляют лишь некоторые ферменты, действующие в экстремальных условиях, например, пепсин, находящийся в кислом желудочном соке. Активный центр состоит из зоны связывания и каталитической зоны. При этом субстрат по конфигурации должен соответствовать зоне связывания (см. рис.). При этом молекула субстрата может взаимодействовать с разными функциональными группами фермента (R1……R163). Зона связывания узнает “свой” субстрат, что определяется пространственным соответствием. Изменения в субстрате, которые позволяют ему подходить к ферменту «как ключ - замок», определяются каталитической зоной. В ней есть высоко активные группы (тиогруппа – тиоловые ферменты, соответственно те вещества, которые связывают эти группы называются - тиоловые яды; гидроксогруппы из серина – сериновые яды; карбоксильные, аминогруппы и так далее) Все эти группы соответсвуют изменению конфигурации субстрата. В момент сближения фермента и субстрата происходит изменение конфигурации каталитической зоны фермента под влиянием истинного субстрата. Теорию образования фермент-субстратного комплекса предложил Э. Фишер, который утверждал, что фермент и субстрат изначально подходят друг к другу как «ключ и замок». Более сответствует действительности теория Кошланда, согласно которой субстрат как бы «подстраивает» каталитическую зону под себя. Если субстрат не вызывает изменение каталитической зоны, то нет и продукта реакции, то-есть не образуется фермент-субстратный комплекс при вхождении в активный центр, субстрат жестко ориентирован, что и вызывает увеличение стерического коэффициента. Ферменты повышают скорости катализируемых ими реакций в 108 – 1012 раз. Например, уреаза ускоряет гидролиз мочевины в десять в четырнадцатой степени раз при pH =8 и 20 градусах по Цельсию. Существует четыре основных фактора определяющих способность ферментов ускорять химические реакции:
1) сближение и ориентация субстрата по отношению к каталитической группе. То-есть фермент способен связывать молекулу субстрата таким образом, что атакуемая ферментом связь оказывается не только расположенной в непосредственнрй близости от каталитической группы, но и правильно ориентируемой по отношению к ней. В результате этого вероятность того, что фермент-субстратный комплекс достигнет переходного состояния значительно увеличивается.
2) Напряжение и деформация с чувствительной к действию фермента связи, возникающие в следствие индуцированного соответствия между молекулами субстрата и фермента. То-есть присоединение субстрата может вызвать конформационные изменения в молекуле фермента, которые приводят к напряжению структуры активного центра, а также несколько деформируют связанный субстрат, облегчая тем самым достижение фермент-субстратным комплексом «переходного состояния». При этом возникает так называемая «индуцированное соответствие» фермента субстрату. Таким образом, небольшие изменения третичной или четвертичной структуры относительно крупной молекулы фермента могут приводить к деформации молекулы субстрата.
3) Общий кислотно-основной катализ. В активном центре фермента могут находиться группы специфических аминокислотных остатков, которые являются хорошими донорами (карбоксильная, NH3+, тиогруппа и др.) или акцепторами протонов (- СОО-, аминогруппа, -S-, и так далее). Такие кислотные или основные группы реакции, причем значительно быстрее, чем в случае не катализируемых реакций.
4) Ковалентный катализ. Ферменты реагируют со своим субстратами, образуя очень нестабильные, ковалентно связанный фермент-субстратные комплексы, из которых в ходе последующих реакций образуются продукты
СВОЙСТВА ФЕРМЕНТОВ.
Ферменты харктеризуются очень высокой активностью, гораздо большей, чем обычные катализаторы. Наример одна молекула каталазы способна за одну минуту расщепить 5000000 молекул перекиси водорода. Одна молекула карбангидразы за одну минуту катализирует взаимодействие 36000000 молей углекислого газа в реакции: СО2 + Н2О = Н2СО3. Молярная активность большинства ферментов оценивается превращением 1000 – 10000 молей. Но даже такие значения говорят о существенном ускорении реакции, если принять во внимание, что для большинства органических реакций в отсутствии катализатора даже при нагревании требуются минуты и часы. В отличие от неорганических катализаторв, ферменты обладают специфичностью. Это определяется соответствием конфигурации активного центра фермента и субстрата. Различают следующие виды специфичности:
1) Абсолютная – фермент действует только на одно определенное вещество. Например, L-аргиназа расщепляет только L-аргинин. Так действуют большинство дегидрогеназ (ЛДГ, и т. д.).
2) Абсолютная групповая – фермент вызывает один и тот же каталитический эффект у группы соединений (как правило, у соединений одного гомологического ряда). Например, алкогольдегидрогеназа – окисляет этиловый, пропиловый, бутиловый и т. д. спирты.
3) Относительная групповая – идет катализ разных классов органических веществ. Например, трипсин прояввляет пептидазную активность в белках и эстеразную в сложных эфирах.
4) Стереохимическая – расщипляется только определенная форма изомеров (D-L, альфа–бета, цис – транс и т. д.). Активность ферментов в значительной мере зависит от рН. При этом для работы каждого фермента характерен свой оптимум рН. Например,пепсин наиболее активен при рН = 1 – 2, амилаза – при рН = 7; аминокислотоксидаза – при рН = 10 – 11. Чувствительность к изменеию рН объясняется следующим образом: при изменении реакции среды изменяется тип ионизации функциональных групп ферментов, в результате этого образуются новые ионные связи, или разрушаются старые, и, как следствие, изменяется конформация молекулы белка-фермента. Это в свою очередь приводит к изменению физико-химических свойств и биологической активности. В клинике при такой ситуации вводятся буферные растворы, которые способствуют нормализации рН, соответственно восстанавливается и активность фермента. Например, при уменьшении кислотности желудочного сока вводится раствор соляной кислоты, что приводит к восстановлению активности пепсина.
Ферменты термолабильнны. При увеличении температуры на каждые 10 градусов скорость реакции увеличивается в 2 – 4 раза. Оптимальная температура для работы фермента 37 – 40 градусов по Цельсию. При дальнейшем повышении температуры ферменты, являясь белками, подвергаются денатурации, теряя ферментативную активность. В этом случае денатурация будет необратима. При снижении температуры ферменты тоже теряют свою активность, но это явление обратимое. Это свойство используется в криохирургии, чтобы в тканях замедлить все процессы, чтобы запасов кислорода и энергии в тканях хватило на более длительное время; для хранения ферментов.
Ферменты фотолабильны. Они очень чувствительны к ультрафиолетовым лучам, так как ферменты – это белки и они при действии ультрафиолета денатурируют (это лежит в основе ультрафиолетового облучения в медицине).
Кинетика – это раздел физической химии, изучающий скорости протекания реакций. Кинетические исследования лежат в основе понимания механизма реакции: во сколько стадий протекает химическая реакция, какова природа превращений на каждой стадии, какая стадия самая медленная, то-есть определяющая скорость суммарной реакции. Описание реакций в таких понятиях и называется механизмом реакции. Хотя скорости реакций определяются экспериментально, соотношение между скоростью и концентрациями реагентов можно выразить в виде простого уравнения. При записи таких уравнений используется константа скорости К. При постоянной температуре К – это постоянная, характеризующая быстроту протекания реакции. Большое значение для объяснния ферментативной кинетики имели работы Михаэлиса и Ментена. Они работали с дрожжевым экстрактом, содержащим инвертазу: сахароза = глюкоза + фруктоза (реакция идет под действием инвертазы). В одной серии опытов [S] = const, а количество фермента изменялось (отмечалась линейная зависимость), во второй – [F] = const, а [S] – изменялась (увеличивалась), при этом отмечалась нелинейная гиперболическая зависимость скорости от [S]. Для обьяснения полученных результатов предполагалось образование фермент- субстратного комплекса.
 K1 K3
K1 K3
F + S FS P + F
 K2
K2
 |
V1 = K1 [F] [S]
V2 = K2 [FS] На основании закона действующих масс.
V3 = K3 [FS]
Наиболее важна V3, так как дает продукт реакции. Но [FS] определить практически невозможно. Используют суммарное уравнение Михаэлиса – Ментена:
K3 [F0] [S] K2 + K3

 \/ = Km =
\/ = Km =
Km + [S], где K1
V3 – скорость приводящая к образованию продукта.
К3 – константа этой скорости.
[F0] – сумма фермента в свободном состоянии и в составе фермент-субстратного комплекса ([F] + [FS]).
[S] – концентрация субстрата.
Кm – константа Михаэлиса.
Модель фермент-субстратного комплекса, построенная на основе лабораторных методов определения V3 при изменении [S] и при [F] = константа и приводящая к кинетическому уравнению Михаэлиса – Ментена и составляет основу ферментативной кинетики.
Константа Михаэлиса и максимальная скорость – кинетические параметры, отражающие механизм действия фермента, так что они являются основой для понимания того, как действует фермент. Еще большую информацию дают изменения константы Михаэлиса и (или) максимальной скорости, полученной вследствии изменений условий реакций, наличие другого субстрата или благодаря обработки фермента, приводящей к изменению его структуры.
Значение константы Михаэлиса и максимальной скорости находят путем анализа кинетических данных. Проанализируем уравнение Михаэлиса – Ментона:
1)Концентрация субстрата постоянна и намного больше концентрации фермента. В таком случае скорость реакции будет зависеть только от концентрации фермента и, если концентрация фермента будет приближаться к концентрации субстрата, то скорость реакции замедлится.

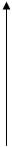 V3
V3
 |
[F0]
2) Допустим, что концентрация фермента постоянна и достаточно высока. Проанализируем в этих условиях уравнение Михаэлиса – Ментона:
а) Концентрация фермента намного больше константы Михаэлиса. Тогда в знаменателе [S] мы можем пренебречь; К3, константа Михаэлиса и [F0] – величины постоянные и выходит что скорость реакции зависит прямо пропорционально только от концентрации субстрата в числителе. Но это справедливо только для малых концентраций субстрата, что мы обговорим в предварительных условиях.
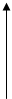 V3
V3
 |
 [S]
[S]
б) Концентрация субстрата намного больше константы Михаэлиса. Тогда в знаменателе мы можем пренебречь константой Михаэлиса, концентрации субстрата в числителе и в знаменателе сокращаются и получается, что при значительных концентрациях субстрата скорость реакции от концентрации субстрата не зависит. Такую скорость называют максимальной (Vmax).
Vmax * [S]
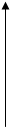
 V3 Следовательно: V3 =
V3 Следовательно: V3 =
Km + [S]
Так как полное насыщение фермента субстратом, то
Vmax = K3 * [FS] = K3 * [E0], то-есть [E0] = [FS],
Vmax так как [E] = 0

 [S]
[S]
При достижении максимльной скорости происходит полное насыщение фермента субстратом. Максимальная скорость характеризует каталитическую силу фермента, а также активирующий эффект активатора и силу ингибитора. То-есть, по сути, максимальная скорость выражает эффективность действия фермента. Для сравнения каталитической активности различных ферментов максимальную скорость выражают через количество (в молях) каждого фермента. Такое преобразование приводит к величине, которую называют молярной активностью. Она выражается числом молей субстрата, реагирующего с одним молем фермента за единицу времени (за одну минуту или секунду). Например, карбангидраз, каталаза и так далее. Активнрсть фермента так же можно выражать в единицах ферментативной активности. Одна энергия катализирует превращение субстрата со скоростью 1мкмоль в минуту. Удельная активность – это ферментативная активность, отнесенная на 1 мг белка. Мы уже упоминали, что Км = (К2 + К3) / К1. Но, К1,2,3 нельзя определить практически, так как не известно сколько существует [FS]. Константу Михаэлиса можно рассчитать графически. Допустим, что \/3 = ½ \/мах, то-есть при 50% насыщения фермента субстратом, тогда \/мах/2=(\/мах х [S]) /(Km + [S]), то-есть 2[S]=Км+[S] Получается, что константа Михаэлиса численно равна концентрации субстрата, при которой скорость ферментативной реакции равна ½ максимальной скорости. При этом связывается половина имеющегося фермента. Константу Михаэлиса выражают в единицах концентрции (моль). Для одномолекулярных реакций константа Михаэлиса равна десять в минус второй – десять в минус пятой степени моль.
Ферменты могут иметь одинаковые каталитические силы, но разные константы Михаэлиса. Константа Михаэлиса характеризует специфичность фермента к субстрату. Чем меньше константа Михаэлиса, тем выше специфичность, тем быстрее образуется фермент-субстратный комплекс, или фермент-субстратный комплекс быстрее распадается на продукт и фермент, или и то и другое. Активаторы и ингибиторы влияют на константу Михаэлиса и максимальную скорость.
Пример, гексакиназа (фермент, катализирующий превращение простых сахаров в фосфоэфиры). Для ее насыщения субстратом на 50% глюкозы требуется в 1000 раз меньше, чем аллозы.
О = С – Н О = С – Н
| |
Н – С – ОН Н – С - ОН
| |
HO – C – H H – C – OH
| |
H – C - OH H – C – OH
| |
H – C – OH H – C – OH
| |
CH2OH CH2OH


 Глюкоза аллоза
Глюкоза аллоза

\/3

- \/мах -| - - - - - - - - |- - - -
|
_ _ _ |_ _ _ _ _ _ |
| |
| |
| |
| | [S]
Км Км2
Из этих дух графиков видно, что из этих двух ферментов более специфичен тот, который имеет меньшую величину константы Михаэлиса, так как в этих условиях быстрее достигается максимальная скорость реакции. Определить константу Михаэлиса можно и используя уравнение обратных величин (рассчитанное Лайнуивером - Бэрком), на основании которого строится одноименный график:
1 / \/3 Этот метод называеися методом двойных обратных величин
и основан на алгебраическом преобразовании исходного
уравнения гиперболы по Михаэлису-Ментону к уравнению
прямой.
1 / \/мах

- 1 / Км 1[S]
ИНГИБИТОРЫ ФЕРМЕНТОВ.
Активность ферментов зависит от действия активаторов и ингибторов ферментов. Соответственно, ингибиторы – это вещества, действие которых снижает активность ферментов, а активаторы – вещества, при действии которых активность ферментов повышается. Различают по специфичности: специфические (антитрипсин) и неспецифические; по месту действия: на активный центр, аллостерические.
Антибиотики, ативирусные агенты, противоопухолевые средства, инсектециды и гербициды – о существовании этих средств теперь знают все. Действие многих из них основаго на способности ингибировать (то-есть затормаживать) работу клеточных ферментов, обычно какого-то определенного фермента. Ингибирование ферментов происходит и в природе, представляя собой одну из систем биорегуляции. Ферменты никогда не работают монотонно. Их активность постоянно меняется и в значительной мере зависит от постоянно меняющихся условий внешней и внутренней среды.

 Различают обратимое ингибирование: ингибитор + фермент (ингибитор – фермент) и необратимое ингибирование: ингибитор + фермент (ингибитор-фермент). В свою очередь обратимые ингибирование делят на конкурентное и неконкурентное. Конкурентное ингибирование основано на близости структур молекул ингибитора и субстрата, то-есть возникает конкуренция за фермент, за его активный центр. Например, субстратом для действия фермента сукцинатдегидрогеназы является янтарная кислота:
Различают обратимое ингибирование: ингибитор + фермент (ингибитор – фермент) и необратимое ингибирование: ингибитор + фермент (ингибитор-фермент). В свою очередь обратимые ингибирование делят на конкурентное и неконкурентное. Конкурентное ингибирование основано на близости структур молекул ингибитора и субстрата, то-есть возникает конкуренция за фермент, за его активный центр. Например, субстратом для действия фермента сукцинатдегидрогеназы является янтарная кислота:
СООН СООН
| |
СН2 - H2 СН F – сукцинатдегидрогеназа.
 | ||
| ||
СН2 F СН
| |
СООН СООН
Янтарная Фумаровая
кислота кислота
Сильнейшим ингибитором этого фермента является малоновая кислота (СООН – СН2 - СООН): образуется комплекс (фермент-ингибитор), который не дает продукта реакции, так как не может идти дегидрирование. При этом уменьшается число молекул свободного фермета, способного взаимодействовать с субстратом и скорость понижается. Но этот вид ингибирования обратим, его можно пресечь постепенным насыщением среды субстратом, то-есть увеличением его концентрации, то-есть мы можем сделать вывод, что при конкурентном ингибировании существует прямая зависимость от концентраций субстрата и ингибитора. При конкурентном ингибировании максимальная скорость постоянна, но константа Михаэлиса увеличивается (то-есть снижается специфичность) и для достижения максимальной скорости нужно повышать концентрацию субстрата.

 1 / \/3 не конкурентное ингибирование
1 / \/3 не конкурентное ингибирование
 -- конкурентное ингибирование
-- конкурентное ингибирование
 без ингибирования
без ингибирования

1 / Км 1 / [S]
Зная принципы конкурентного ингибирования, можно искусственно находить (например, синтезировать) ингибиторы ферментов, что и используется на практике. Например, парааминобензойная кислота используется для синтеза фолиевой кислоты, необходимой для жизнедеятельности микроорганизмов. Ингибитором фермента, катализирующего эту реакцию является сульфаниловая кислота, которая входит в состав сульфаниламидных препаратов (сульфадиметоксин, сульфадимезин и т. д.). Введение этих препаратов ингибирует синтез фолиевой кислоты, не давая возможности жить и развиваться микроорганизмам.
COOH SO3H O

 | | || N
| | || N






 HN __R
HN __R
 1 2 3
1 2 3


 H2N
H2N




 | |
| |
NH2 NH2
1 – парааминобензойная кислота; 2 – сульфониловая кислота; 3 – фоливая кислота.
Тетрогидрофоливая кислота – это очень важный ко-фермент нескольких ферментов, участвующих в синтезе ДНК и РНК. При введении сульфаниламидных препаратов, немедленно наблюдается ингибирование фермента, катализирующего утилизацию парааминобензойной кислоты (ПАБК) при синтезе фолиевой кислоты. Пониженный уровень фолиевой кислоты приводит к уменьшению синтеза нуклеиновых кислот.
Окончательным результатом этих событий является гибель микроорганизмов. Или, на основе урацила получен препарат 5 – фторурацил, который используется для лечения опухолей, так как блокирует синтез нуклеиновых кислот.
О
| |



 HN -- F 5 – фторурацил.
HN -- F 5 – фторурацил.
O
NH
Аллопуринол (применяется при лечении конкурентного ингибирования). В некоторых случаях конкурентным ингибированием можно объяснить ингибирующие действие конечного продукта на фермент,участвующий в синтезе этого продукта. Такой тип биорегуляции часто встречается, его называют – ингибирование конечным продуктом.
Неконкурентные ингибиторы взаимодействуют не со всем активным центром, а с отделными функциональными группами в каталитической зоне. Такие ингибиторы имееют химическое сродство к какой-либо функциональной группе. Например, ионы серебра, ртути являются тиоловыми ядами, то-есть блокируют тиогруппы. Цианиды – соли синильной кислоты, являются дыхательными ядами, они связывают двух- и трехвалентное железо в составе простетической группы сложных белков-ферментов. Могут связываться и другие группы (карбоксильная, гидроксо- и т.д.). Притаком виде ингибировании повышение концентрации субстрата (чтобы прекратить ингибирование) не эффективно. Этот вид ингибирования можно убрать, вытесняя ингибитор из фермента. Например, в случае действия тиоловых ядов, добавляют вещества, имеющие тиогруппу (тиолы), например, цистеин. При этом виде ингибирования константа Михаэлиса является постоянной, то-есть специфичность не меняется, но уменьшается максимальная скорость, то-есть снижается каталитическая сила. К такого рода ингибиторам относятся: иприт, люизит. Снижение каталитической силы фермента (то-есть уменьшение максимальной скорости) при неизменной константе Михаэлиса возможно в случае, когда субстрат имеет одинаковое сродство к ферменту и фермент-ингибиторному комплексу, а ингибитор – к ферменту и к фермент-субстратному комплексу. Если сродство субстрата и ингибитора к ферменту и соответствующему комплексу различно (а чаще бывает, что так и есть), то меняются и максимальная скорость и константа Михаэлиса. Это во многих случаях характерно для ингибирования конечными продуктами.
НЕКОНКУРЕНТНОЕ ИНГИБИРОВАНИЕ может быть и необратимым (ингибитор ковалентно связывается с ферментом или фермент-субстратным комплексом, необратимо меняя нативную конформацию). При этом виде ингибирования между ингибитором и ферментом существует очень высокое сродство. Например, монойодуксусная кислота (СН2 I – СООН), необратимо блокируют тиогруппы за счет связи атома йода с атомом водорода, а атом серы прочно связывается с остатком – СН2СООН. При этом добавление тиолов не помогает, то-есть ингибирование является необратимым. Или например, ФОС (фосфоорганические соединения) блокируют многие ферменты (например, эстеразы и другие), содержащие серин, образуя очень прочную связь. Это свойство используют в изготовлении БОВ (боевых отравляющих веществ: табун, зарин, зоман, \/-газы) и в сельском хозяйстве для борьбы с вредителями. Из лекарственных препаратов, действие которых основано на необратимом ингибировании, можно назвать пенициллин, который ингибирует действие одного из ферментов участвующих в сборке клеточной стенки бактерий. Клетки, не имеющие клеточной стенки (их называют протопласты) легко лизируются. Интенсивное и неосторожное использование антибиотиков привело к быстрой эволюции бактериальных штаммов, устойчивых к антибиотикам. Поэтому очень важен поиск и синтез новых антибиотиков, против которых бактерии еще не имеют устойчивости. Например, в 1981году открыт новый класс антибиотиков, монобактами, по структуре сходных с пенициллином. Еще один пример лекарственного средства, действие которого основано на ковалентной модификации фермента – аспирин.
Существует и бесконкурентное ингибирование (его можно рассматривать как разновидность не конкурнтного) – когда ингибитор связывается не с ферментом а с фермент-субстратным комплексом, образуя комплекс фермент-субстрат-ингибитор. Здесь всегда действие фермента будет аллостерическим, так как активный центр занят субстратом, и ингибитор присоединяется к ферменту не к активному центру, а к аллостерическим зонам, удаленных от активного центра. Это приводит к изменению конформации аллостерической зоны, кооперативно с ней меняется и структура активного цента, то-естьменяется структура всей молекулы, приводя к тому что комплекс фермент-субстрат-иннгибитор не распадается и не дает продукта реакции. При таком виде ингибирования меняется (уменьшается), специфичность и каталитическая сила фермента.
АКТИВАТОРЫ ФЕРМЕНТОВ.
Активаторы повышают активность ферментов в десятки, сотни раз. Активаторы бывают специфические и неспецифические. Например, хлорид натрия для амилазы специфический, ионы магния, марганец и цинк – неспецифические (есть целая группа: марганец-, магнийзависимых ферментов). Некоторые вещества для одних ферментов являются активаторами, а для других – ингибиторами (например, ионы меди для полифенолоксидазы – активатор, а для амилазы – ингибитор.