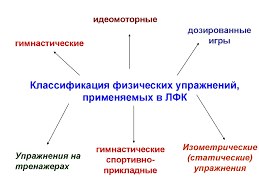
2020-04-20
2020-04-20 863
863
15 октября 1980 г. на Нью-Йоркской фондовой бирже произошло знаменательное событие: уже через 20 минут после начала торгов стоимость одной акции биотехнологической компании Genentech поднялась с 35 до 89 долларов. Это был рекордный для того времени скачок цен на акции коммерческого предприятия. К моменту закрытия торгов в тот день цена одной акции Genentech составляла 71,25 доллара, а стоимость всех 528 тысяч акций была столь баснословно высока, что мелкие инвесторы, собиравшиеся приобрести небольшой пакет акций, не имели никаких шансов.
По-видимому, это был первый случай в истории, когда о начале великой технологической революции возвестил биржевой колокол. В 1980 г., когда фирма Genentech впервые предложила обществу свои акции, это была небольшая компания в Калифорнии, в течение четырех лет успешно работавшая над проблемой получения рекомбинантных ДНК. За два года до этого ученым компании удалось выделить фрагменты гена (последовательности ДНК), кодирующие человеческий инсулин, и перенести их в генетические элементы (клонирующие векторы), способные реплицироваться в клетках обычной кишечной палочки (Escherichia coli). Эти бактериальные клетки работали как биологические фабрики по производству человеческого инсулина, который после соответствующей очистки мог использоваться как лекарственный препарат для больных диабетом, дающих аллергическую реакцию на свиной инсулин. Еще десять лет назад такое развитие событий представлялось нереальным, но сегодня все это стало вполне привычным.
Генная инженерия появилась благодаря работам многих исследователей в разных отраслях биохимии и молекулярной генетики. На протяжении многих лет главным классом макромолекул считали белки. Существовало даже предположение, что гены имеют белковую природу. Лишь в 1944 году Эйвери, Мак Леод и Мак Карти показали, что носителем наследственной информации является ДНК. С этого времени начинается интенсивное изучение нуклеиновых кислот. Спустя десятилетие, в 1953 году Дж. Уотсон и Ф. Крик создали двуспиральную модель ДНК. Именно этот год принято считать годом рождения молекулярной биологии.
На рубеже 50 - 60-х годов были выяснены свойства генетического кода, а к концу 60-х годов его универсальность была подтверждена экспериментально. Шло интенсивное развитие молекулярной генетики, объектами которой стали E. coli, ее вирусы и плазмиды. Были разработаны методы выделения высокоочищенных препаратов неповрежденных молекул ДНК, плазмид и вирусов. ДНК вирусов и плазмид вводили в клетки в биологически активной форме, обеспечивая ее репликацию и экспрессию соответствующих генов. В 70-х годах был открыт ряд ферментов, катализирующих реакции превращения ДНК. Особая роль в развитии методов генной инженерии принадлежит рестриктазам и ДНК-лигазам.
Историю развития генетической инженерии можно условно разделить на три этапа.
Первый этап связан с доказательством принципиальной возможности получения рекомбинантных молекул ДНК in vitro. Эти работы касаются получения гибридов между различными плазмидами. Была доказана возможность создания рекомбинантных молекул с использованием исходных молекул ДНК из различных видов и штаммов бактерий, их жизнеспособность, стабильность и функционирование.
Второй этап связан с началом работ по получению рекомбинантных молекул ДНК между хромосомными генами прокариот и различными плазмидами, доказательством их стабильности и жизнеспособности.
Третий этап - начало работ по включению в векторные молекулы ДНК (ДНК, используемые для переноса генов и способные встраиваться в генетический аппарат клетки-рецепиента) генов эукариот, главным образом, животных. Формально датой рождения генетической инженерии следует считать 1972 год, когда в Стенфордском университете П. Берг, С. Коэн, Х. Бойер с сотрудниками создали первую рекомбинантную ДНК, содержавшую фрагменты ДНК вируса SV40, бактериофага и E. coli.
Возможности генной инженерии. Важной составной частью биотехнологии является генетическая инженерия. Родившись в начале 70-х годов, она добилась сегодня больших успехов. Методы генной инженерии преобразуют клетки бактерий, дрожжей и млекопитающих в "фабрики" для масштабного производства любого белка. Это дает возможность детально анализировать структуру и функции белков и использовать их в качестве лекарственных средств.
В настоящее время кишечная палочка (E. coli) стала поставщиком таких важных гормонов как инсулин и соматотропин. Ранее инсулин получали из клеток поджелудочной железы животных, поэтому стоимость его была очень высока. Для получения 100 г кристаллического инсулина требуется 800-1000 кг поджелудочной железы, а одна железа коровы весит 200 - 250 грамм. Это делало инсулин дорогим и труднодоступным для широкого круга диабетиков. В 1978 году исследователи из компании "Генентек" впервые получили инсулин в специально сконструированном штамме кишечной палочки. Инсулин состоит из двух полипептидных цепей А и В длиной 20 и 30 аминокислот. При соединении их дисульфидными связями образуется нативный двухцепочечный инсулин. Было показано, что он не содержит белков E. coli, эндотоксинов и других примесей, не дает побочных эффектов, как инсулин животных, а по биологической активности от него не отличается. Впоследствии в клетках E. coli был осуществлен синтез проинсулина, для чего на матрице РНК с помощью обратной транскриптазы синтезировали ее ДНК-копию. После очистки полученного проинсулина его расщепили и получили нативный инсулин, при этом этапы экстракции и выделения гормона были сведены к минимуму. Из 1000 литров культуральной жидкости можно получать до 200 граммов гормона, что эквивалентно количеству инсулина, выделяемого из 1600 кг поджелудочной железы свиньи или коровы.
Соматотропин - гормон роста человека, секретируемый гипофизом. Недостаток этого гормона приводит к гипофизарной карликовости. Если вводить соматотропин в дозах 10 мг на кг веса три раза в неделю, то за год ребенок, страдающий от его недостатка, может подрасти на 6 см. Ранее его получали из трупного материала, из одного трупа: 4 - 6 мг соматотропина в пересчете на конечный фармацевтический препарат. Таким образом, доступные количества гормона были ограничены, кроме того, гормон, получаемый этим способом, был неоднороден и мог содержать медленно развивающиеся вирусы. Компания "Genentec" в 1980 году разработала технологию производства соматотропина с помощью бактерий, который был лишен перечисленных недостатков. В 1982 году гормон роста человека был получен в культуре E. coli и животных клеток в институте Пастера во Франции, а с 1984 года начато промышленное производство инсулина и в СССР. При производстве интерферона используют как E. coli, S. cerevisae (дрожжи), так и культуру фибробластов или трансформированных лейкоцитов. Аналогичными методами получают также безопасные и дешевые вакцины.
На технологии рекомбинантных ДНК основано получение высокоспецифичных ДНК-зондов, с помощью которых изучают экспрессию генов в тканях, локализацию генов в хромосомах, выявляют гены, обладающие родственными функциями (например, у человека и курицы). ДНК-зонды также используются в диагностике различных заболеваний.
Технология рекомбинантных ДНК сделала возможным нетрадиционный подход "белок-ген", получивший название "обратная генетика". При таком подходе из клетки выделяют белок, клонируют ген этого белка, модифицируют его, создавая мутантный ген, кодирующий измененную форму белка. Полученный ген вводят в клетку. Если он экспрессируется, несущая его клетка и ее потомки будут синтезировать измененный белок. Таким образом можно исправлять дефектные гены и лечить наследственные заболевания.
Если гибридную ДНК ввести в оплодотворенное яйцеклетку, могут быть получены трансгенные организмы, экспрессирующие мутантный ген и передающие его потомками. Генетическая трансформация животных позволяет установить роль отдельных генов и их белковых продуктов как в регуляции активности других генов, так и при различных патологических процессах. С помощью генетической инженерии созданы линии животных, устойчивых к вирусным заболеваниям, а также породы животных с полезными для человека признаками.
Стратегия переноса функциональной единицы наследственности (гена) из одного организма в другой была разработана американскими учеными Стэнли Коэном и Гербертом Бойером в 1973 г. И Коэну, и Бойеру, и многим другим было ясно, что технология рекомбинантных ДНК предоставляет огромные возможности. Как в то время отмечал Коэн, «...есть надежда, что удастся ввести в [бактериальную клетку] E. coli гены, ассоциированные с метаболическими или синтетическими функциями, присущими другим биологическим видам, например гены фотосинтеза или продукции антибиотиков».
Однако одним из первых откликов научного мира на создание новой технологии был мораторий на некоторые биотехнологические эксперименты, считавшиеся потенциально опасными. Запрет на собственные исследования был провозглашен группой молекулярных биологов, включая Коэна и Бойера. Они считали, что объединение генов, происходящих из двух разных организмов, может случайно привести к созданию нового организма с нежелательными и опасными свойствами. Прошло несколько лет, у ученых накопился опыт работы с новой технологией, были согласованы инструкции по обеспечению безопасности этих работ, и страсти постепенно улеглись. Временное прекращение реализации некоторых научных проектов, связанных с рекомбинантными ДНК, не уменьшило энтузиазма генных инженеров. Новая технология продолжает привлекать беспрецедентное внимание как со стороны общественности, так и со стороны ученых.
Весть о клонировании генов, осуществленном Коэном и Бойером, облетела весь мир. Многие исследователи немедленно оценили все преимущества этой стратегии и создали огромное количество методик, следуя которым, можно было с высокой эффективностью и относительно просто идентифицировать, выделять, охарактеризовывать и использовать гены. Эти технологические разработки внесли значительный вклад в развитие практически всех биологических дисциплин, включая науку о поведении животных, биологию развития, молекулярную эволюцию, клеточную биологию и генетику человека, однако наиболее глубокие изменения произошли в области биотехнологии.
Методы генной инженерии. Генетическая инженерия - получение новых комбинаций генетического материала путем проводимых вне клетки манипуляций с молекулами нуклеиновых кислот и переноса созданных конструкций генов в живой организм, в результате которого достигается их включение и активность в этом организме и у его потомства. Речь идет о направленном, по заранее заданной программе конструировании молекулярных генетических систем вне организма с последующим введением их в живой организм. При этом рекомбинантные ДНК становятся составной частью генетического аппарата рецепиентного организма и сообщают ему новые уникальные генетические, биохимические, а затем и физиологические свойства.
Цель прикладной генетической инженерии заключается в конструировании таких рекомбинантных молекул ДНК, которые при внедрении в генетический аппарат придавали бы организму свойства, полезные для человека. Например, получение «биологических реакторов» - микроорганизмов, растений и животных, продуцирующих фармакологически значимые для человека вещества, создание сортов растений и пород животных с определёнными ценными для человека признаками.
Методы генной инженерии позволяют провести генетическую паспортизацию, диагностировать генетические заболевания, создавать ДНК-вакцины, проводить генотерапию различных заболеваний.
Технология рекомбинантных ДНК использует следующие методы:
1. специфическое расщепление ДНК рестрицирующими нуклеазами, ускоряющее выделение и манипуляции с отдельными генами;
2. быстрое секвенирование всех нуклеотидов очищенном фрагменте ДНК, что позволяет определить границы гена и аминокислотную последовательность, кодируемую им;
3. конструирование рекомбинантной ДНК;
4. гибридизация нуклеиновых кислот, позволяющая выявлять специфические последовательности РНК или ДНК с большей точностью и чувствительностью, основанную на их способности связывать комплементарные последовательности нуклеиновых кислот;
5. клонирование ДНК: амплификация in vitro с помощью цепной полимеразной реакции или введение фрагмента ДНК в бактериальную клетку, которая после такой трансформации воспроизводит этот фрагмент в миллионах копий;
6. введение рекомбинантной ДНК в клетки или организмы.
Ферменты, применяемые при конструировании рекомбинантных ДНК, можно разделить на несколько групп:
- ферменты, с помощью которых получают фрагменты ДНК (рестриктазы);
-ферменты, синтезирующие ДНК на матрице ДНК (полимеразы) или РНК (обратные транскриптазы);
- ферменты, соединяющие фрагменты ДНК (лигазы);
- ферменты, позволяющие осуществить изменение структуры концов фрагментов ДНК.
Рестриктазы. Рестриктазы (рестрицирующие эндонуклеазы, эндонуклеазы рестрикции) - это ферменты, узнающие и атакующие определенные последовательности нуклеотидов в молекуле ДНК (сайты рестрикции).
Еще в 1953 году было обнаружено, что ДНК определенного штамма E. coli, введенная в клетки другого штамма (например, ДНК штамма В - в клетки штамма С) не проявляет, как правило, генетической активности, так как быстро расщепляется на мелкие фрагменты.
В 1966 году было показано, что это явление связано со специфической модификацией хозяйской ДНК - она содержит несколько метилированных оснований, отсутствующих в немодифицированной ДНК, причем метилирование (добавление к основанию метильной группы) происходит уже после завершения репликации. Бактерия способна отличить свою собственную ДНК от любой вторгающейся «чужеродной» именно по типу ее модификации. За «метку» отвечают метилирующие ферменты модификации, так называемые ДНК-метилазы. Различие в модификации делает чужеродную ДНК чувствительной к действию рестрицирующих ферментов, которые узнают отсутствие метильных групп в соответствующих сайтах.
Системы рестрикции и модификации широко распространены у бактерий; их существование играет важную роль в защите резидентной ДНК от загрязнения последовательностями чужеродного происхождения.
Рестриктаза, которая расщепляла неметилированную ДНК была выделена в 1968 г. Мезельсоном и Юанем. Этот фермент был высокоспецифичен по отношению к определенной последовательности ДНК, но расщеплял молекулы неспецифически, в другом месте, на некотором удалении от участка узнавания. Вскоре, в 1970 г. Смит и Вилькокс выделили из Haemophilus influenzae первую рестриктазу, которая расщепляла строго определенную последовательность ДНК (Hind III). Поскольку разные бактерии по-разному метят свою ДНК, то и рестриктазы должны узнавать разные последовательности. И действительно, с тех пор выделены рестриктазы, узнающие более 150 сайтов рестрикции (мест расщепления ДНК).
Многие рестриктазы вносят разрывы в две цепи ДНК со смещением на несколько нуклеотидов и образованием на концах фрагменте! коротких одноцепочечных участков. Они способны образовывать комплементарные пары оснований с любым другим одноцепо- чечным участком, полученным с помощью того же фермента (липкие концы). Липкие концы позволяют легко соединить два любы;, фрагмента ДНК в одно целое. Полученный фрагмент ДНК (любого происхождения) можно встроить в очищенную ДНК плазмиды или бактериального вируса.
Сравнение размеров фрагментов ДНК после обработки соответствующего участка генома набором рестриктаз позволяет построить рестрикционную карту, отражающую расположение определенной последовательности нуклеотидов в данном участке. Сравнением таких карт можно оценить степень гомологии межд\ отдельными генами (участками) без определения их нуклеотидной последовательности. Рестрикционные карты важны для клонирования ДНК, решения эволюционных и филогенетических задач.
В качестве мишеней (мест узнавания) часто выступают палиндромы из 4-6 пар оснований - сайты рестрикции. Точки узнавания рестриктазами симметричны относительно поворота на 180оС, то есть последовательность нуклеотидов слева направо в одной нити такая же, как справа налево в другой. Симметрия подразумевает, что те из них, которые должны быть метилированы, встречаются на обеих цепях ДНК. В результате сайт-мишень может быть полностью метилирован (обе цепи модифицированы), полуметилирован (только одна цепь метилирована) или не метилирован.
Полностью метилированный сайт не подвержен ни рестрикции, ни модификации. Полуметилированный сайт не узнается ферментом рестрикции, но может быть превращен с помощью метилазы в полностью метилированный. У бактерий метилирование, как правило, связано с сохранением имеющегося состояния модификации. Репликация полностью метилированной ДНК ведет к образованию полуметилированной ДНК. Вероятно узнавание полуметилированных сайтов представляет собой обычный этап функционирования метилазы in vivo.
Неметилированный сайт-мишень представляет собой субстрат либо для рестрикции, либо для модификации in vitro. В клетке немодифицированная ДНК с большей вероятностью рестрицируется. Реакция разрезания осуществляется в две ступени. Сначала разрезается одна цепь ДНК, а затем рядом разрезается другая. В областях, прилегающих с каждой стороны к сайту разрезания, может иметь место экзонуклеотическая деградация. Происходит эффективный гидролиз АТФ, роль которого еще не выяснена.
Каким образом фермент узнает один сайт, а разрезает другой, достаточно удаленный? Важно отметить, что белок никогда не отделяется от молекулы ДНК, с которой он первоначально связался. Если фермент инкубировать со смесью модифицированной и немодифицированной ДНК, он предпочтительно разрезает немодифицированную ДНК. Следовательно, узнавая сайт связывания, белок не отделяется от неметилированной ДНК для того, чтобы найти сайт разрезания.
Существуют две альтернативные модели, объясняющие взаимосвязь между сайтами узнавания и разрезания: в соответствии с одной из них движется фермент, согласно другой модели, перемещается ДНК. Если движется фермент, то его перемещение вдоль ДНК будет продолжаться до тех пор, пока он не сделает выбор сайта разрезания. Если же движется ДНК, то фермент остается прикрепленным в сайте узнавания, а ДНК протаскивается через второй сайт связывания на ферменте, и это продолжается до тех пор, пока фермент не достигает области разрезания (пока не охарактеризованной). Получены электронно-микроскопические данные, свидетельствующие, что фермент вызывает образование петли в ДНК и остается, по-видимому, связанным с сайтом узнавания после разрезания; эти данные подтверждают вторую модель.
Полимеразы. Впервые ДНК-полимераза была выделена Корнбергом с сотрудниками в 1958 году из E. coli. ДНК-полимераза I E. coli (Pol I) не связывается с молекулами двухцепочечной кольцевой ДНК. Однако, если такие молекулы денатурировать и получить одноцепочечные формы, то с последними полимераза связывается в количествах, пропорциональных длине этих участков — примерно одна молекула на 300 нуклеотидных остатков. Pol l связывается с одноцепочечными участками двойной спирали ДНК, в местах одноцепочечных разрывов с З'-гидроксилом и 5'-фосфатом, а также с концами двухцепочечных молекул ДНК.
Фермент состоит из мономерной полипептидной цепи с молекулярной массой 103 кДа и имеет 3-х доменную структуру. Каждый домен обладает своей ферментативной активностью: 5’ - 3’ полимеразной, 3’ - 5’ экзонуклезной, 5’ - 3’ экзонуклеазной.
1. 5'— 3' полимеразная активность. Для реакции необходимо наличие одноцепочечной ДНК-матрицы и комплементарного участку этой цепи фрагмента — праймера (затравки) с З'-ОН концом.
2. 3'- 5' экзонуклеазная активность. Гидролизует одноцепочечную или двухцепочечную ДНК с З'-ОН конца. 3'—5' нуклеаза расщепляет диэфирную связь только в неспаренных участках ДНК. Известно, что при полимеразной реакции с определенной частотой возможно включение в растущую цепь некомплементарного нуклеотида. Однако полимераза не может присоединять нуклеотид к неправильно спаренному концу, образовавшемуся при ее участии. На помощь приходит 3'—5' экзонуклеаза, убирающая ошибочный нуклеотид, на место которого затем присоединяется правильный нуклеотид-предшественник. 3'—5' экзонуклеолитическая активность проявляется в направлении, обратном синтезу ДНК (см. рис. 34). Таким образом, 3'—5' экзонуклеазная активность ДНК-полимеразы играет важную роль в точности полимеризации, направляемой матрицей. Эффективность, или число оборотов, данной экзонуклеазы в оптимальных условиях составляет 2% от числа оборотов субъединицы с полимеразной активностью.
3. 5'— 3' экзонуклеазная активность. Деградирует одну цепь двухцепочечной ДНК, начиная со свободного 5'-конца. В отличие от 3'—5' экзонуклеазы 5'—3' экзонуклеаза расщепляет диэфирную связь только в спаренных участках двухцепочечной молекулы ДНК. Более того, в то время как 3'—5' нуклеаза отщепляет одномоментно только один нуклеотид, 5'—3' нуклеаза может вырезать с 5'- конца олигонуклеотиды длиной до десяти остатков (около 20% продуктов гидролиза): Скорость нуклеазного отщепления увеличивается на порядок при одновременно протекающей реакции полимеризации. При этом увеличивается относительное количество олигонуклеотидов в продуктах гидролиза ДНК.
Такое сочетание ферментативных активностей позволяет ДНК-полимеразе I E. coli играть активную роль в репарации повреждений ДНК in vivo. N - концевой домен соединен с соседним петлей из аминокислотных остатков и легко отделяется с помощью протеолитических ферментов. Оставшаяся часть бифункциональна, так как состоит из полимеразы и 3’ - 5’ экзонуклезы. Она названа фрагментом Кленова (по фамилии одного из авторов, описавших ее). Фрагмент Кленова (Pol IK) обычно используют для достройки одноцепочечных 5'-концов на двухцепочечной ДНК, часто генерируемых рестриктазами, до тупых; для синтеза второй цепи на одноцепочечной ДНК, а также для гидролиза одноцепочечных З'-концов на двухцепочечных молекулах ДНК.
Обратная транскриптаза. Обратная транскриптаза используется для транскрипции м-РНК в комплементарную цепь ДНК. При изучении ретровирусов, геном которых представлен молекулами одноцепочечной РНК, было обнаружено, что в процессе внутриклеточного развития ретровирус проходит стадию интеграции своего генома в виде двухцепочечной ДНК в хромосомы клетки-хозяина. В 1964 г. Темин выдвинул гипотезу о существовании вирусспецифичного фермента, способного синтезировать на РНК-матрице комплементарную ДНК. Усилия, направленные на выделение такого фермента, увенчались успехом, и в 1970 г. Темин с Мизутани, а также независимо от них Балтимор открыли искомый фермент в препарате внеклеточных вирионов вируса саркомы Рауса. Данная РНК-зависимая ДНК-полимераза получила название обратная транскриптаза, или ревертаза.
Наиболее детально изучена ревертаза ретровирусов птиц. Каждый вирион содержит около 50 молекул этого фермента. Обратная транскриптаза состоит из двух субъединиц — a (65 кДа) и b (95 кДа), присутствующих в эквимолярном количестве. Обратная транскриптаза обладает, по крайней мере, тремя ферментативными активностями:
1) ДНК-полимеразной, использующей в качестве матрицы как РНК, так и ДНК;
2) активностью РНКазы Н, гидролизующей РНК в составе гибрида РНК - ДНК, но не одно- или двухцепочечную РНК;
3) ДНК-эндонуклеазной активностью.
Первые две активности необходимы для синтеза вирусной ДНК, а эндонуклеаза, по-видимому, важна для интеграции вирусной ДНК в геном клетки-хозяина. Очищенная обратная транскриптаза синтезирует ДНК как на РНК-, так и на ДНК-матрицах. Чтобы начать синтез, ревертазе, как и другим полимеразам, необходим короткий двухцепочечный участок (праймер). Праймером может служить одноцепочечный сегмент как РНК, так и ДНК, которые в процессе реакции оказываются ковалентно связанными с новосинтезированной цепью ДНК.
Обратную транскриптазу преимущественно используют для транскрипции матричной РНК в комплементарную ДНК (кДНК). Реакцию обратной транскрипции проводят в специально подобранных условиях с использованием сильных ингибиторов РНКазной активности. При этом удается получать полноразмерные ДНК-копии целевых молекул РНК. В качестве праймера при обратной транскрипции поли (А)-содержащих мРНК используют олигo (dT), а для молекул РНК, не имеющих З'-поли (А) концов, — химически синтезированные олигонуклеотиды, комплементарные З'-концу изучаемой РНК. После синтеза на мРНК комплементарной цепи ДНК и разрушения РНК (обычно применяют обработку щелочью) осуществляют синтез второй цепи ДНК. При этом используют способность ревертазы образовывать на 3'-концах одноцепочечных кДНК самокомплементарные шпильки, которые могут выполнять функции праймера.
Матрицей служит первая цепь кДНК. Данная реакция может катализироваться как ревертазой, так и ДНК-полимеразой I E. coli. Показано, что сочетание этих двух ферментов позволяет повысить выход полноценных двухцепочечных молекул кДНК. По окончании синтеза первая и вторая цепи кДНК остаются ковалентно связанными петлей шпильки, служившей праймером при синтезе второй цепи. Эту петлю расщепляют эндонуклеазой S1, специфически разрушающей одноцепочечные участки нуклеиновых кислот. Образующиеся при этом концы не всегда оказываются тупыми, и для повышения эффективности последующего клонирования их репарируют до тупых с помощью фрагмента Кленова ДНК-полимеразы I E. coli. Полученную двухцепочечную кДНК можно затем встраивать в клонирующие векторы, размножать в составе гибридных молекул ДНК и использовать для дальнейших исследований.
Лигазы. В 1961 г. Мезельсон и Вейгл на примере фага l показали, что рекомбинация включает разрыв и последующее воссоединение молекул ДНК. Это положило начало поискам фермента, участвующего в сшивании фрагментов ДНК. В 1967 году такой фермент был найден и получил название ДНК-лигаза. Он катализирует синтез фосфодиэфирной связи в 2-х цепочечной молекуле нуклеиновой кислоты.
Иными словами, ДНК-лигазы сшивают рядом расположенные нуклеотиды, образуя связь между остатками сахаров. ДНК-лигазы абсолютно необходимы в процессах репарации ДНК, в процессах репликации - при удвоении цепи ДНК.
В генной инженерии используются 2 типа ДНК-лигаз, отличающихся по потребностям в кофакторах и способу действия. ДНК-лигаза E. coli в качестве кофактора использует дифосфопиридиннуклеотид, а лигаза фага Т4 - АТФ в присутствии Mg2+. Лигаза фага Т4 более универсальна, так как помимо лигирования липких концов способна катализировать реакцию воссоединения двухцепочечных фрагментов ДНК с тупыми концами. Она используется чаще.
Терминальная трансфераза, поли-А – полимераза. Терминальная трансфераза (концевая дезоксинуклеотидилтрансфераза) была обнаружена Боллумом в 1962 году в тимусе теленка. Субстратом терминальной трансферазы при использовании в качестве кофактора ионов Mg2+ является одноцепочечная ДНК с З'-ОН концом или двухцепочечная ДНК с выступающим одноцепочечным З'-ОН концом. Если в качестве кофактора используются Co2+, этот фермент может катализировать присоединение дезоксинуклеотидов к 3’-ОН концу двухцепочечной ДНК с тупыми концами.
При введении в реакцию, направляемую терминальной трансферазой, лишь одного типа дезоксинуклеотидов образуются молекулы ДНК, имеющие гомополимерные 1-цепочечные 3'-концы. Таким же образом можно достроить другим молекулам ДНК гомополимерные 3'-концы, комплементарные первым. Смешение полученных препаратов ДНК при определенных условиях может приводить к формированию гибридных молекул ДНК. Именно с помощью концевой дезоксинуклеотидилтрансферазы в 1972 г. был выполнен первый эксперимент по рекомбинации молекул ДНК in vitro.
Поли (А) - полимераза E. coli была открыта Сиппелом в 1973 году. Она катализирует присоединение к 3’-ОН концу одноцепочечных молекул РНК поли (А) последовательностей. Применяется при подготовке молекул РНК к копированию с них комплементарной ДНК для введения радиоактивной метки в 3’-конец РНК.