2015-06-10
2015-06-10 846
846Прогноз зависит от причины заболевания и степени вовлечения почек. 90% больных с реактивным (вторичным) амилоидозом погибают в течение двух лет с момента установления диагноза, обычно вследствие токсемии, обусловленной хронической инфекцией. У небольшой части больных, проживших достаточно долго, причиной смерти являются вторично-сморщенные амилоидные почки. Эти больные никогда не умирают от печёночной недостаточности.
Продолжительность жизни в группе из 474 больных с AL-амилоидозом составила в среднем 13 мес. Пять лет и более прожили 7%, и только у 1 % больных продолжительность жизни была 10 лет [10]. При наличии застойной сердечной недостаточности выживаемость была хуже.
ЛЕЧЕНИЕ
Лечение амилоидоза подразумевает лечение вызвавшего его заболевания. Так, в случае излечения туберкулёза амилоидоз может регрессировать. Клиническое улучшение при ревматоидном артрите может сопровождаться исчезновением клинических признаков амилоидоза. Специфического лечения не существует.
Во всех случаях семейной средиземноморской лихорадки профилактическое назначение колхицина предотвращает развитие амилоидоза [16].
На AL-амилоид можно воздействовать мелфаланом и преднизолоном, которые уменьшают синтез белков-предшественников [11].
Методом, позволяющим добиться излечения больных с семейной амилоидной нейропатией, является трансплантация печени. Заболевание характеризуется прогрессирующей периферической и вегетативной нейропатией и различной степенью поражения сердца, почек и других органов. В большинстве случаев заболевание обусловлено точечными мутациями генов, кодирующих плазменный транстиретин, синтезируемый печенью. После трансплантации печени аномальные транстиретины исчезают из плазмы и заболевание подвергается частичному обратному развитию. Симптомы вегетативной невропатии в большей степени подвергаются обратному развитию, чем периферической невропатии [14, 15].
Недостаточность a1-антитрипсина [18]
a1-Антитрипсин синтезируется в печени в шероховатой эндоплазматической сети. Он составляет 80—90% всех а,-глобулинов сыворотки, a1-Антитрипсин ингибирует трипсин и другие протеазы. Его недостаточность приводит к повышению активности этих ферментов, особенно эластазы нейтрофилов. Главной мишенью ферментов служат лёгкие, в которых они повреждают альвеолы, что ведёт к развитию эмфиземы.
Ген, кодирующий a1-антитрипсин, локализован на хромосоме 14. В этом локусе существует около 75 различных аллелей, которые можно выделить методом изоэлектрического фокусирования или электрофореза в агарозе в кислой среде либо методом полимеразной цепной реакции. В норме встречается аллель М. Аллели Z и S — наиболее часто встречающиеся патологические аллели, предрасполагающие к возникновению заболевания. От каждого родителя наследуется по одному гену. Результатом их сочетания являются нормальный, промежуточный, низкий или нулевой уровень сывороточного а,-антитрипсина. При генетическом варианте PiMM (ингибитор протеаз) активность сывороточного а,-антитрипсина нормальная и составляет 20—53 мкмоль/л. При варианте PiZZ она снижена до 2,5—7 мкмоль/л, а при Рi00 не выявляется. В обоих последних случаях повышается риск развития эмфиземы. При генетических вариантах PiSS и PiMZ активность а,-антитрипсина составляет 50—60% от нормальной; риск развития заболевания лёгких при этом не повышен. При варианте PiSZ активность a1-антитрипсина снижена до 8—19 мкмоль/л, что сопровождается умеренным риском развития эмфиземы.
Возможны несколько механизмов, посредством которых мутация гена приводит к недостаточности циркулирующего а,-антитрипсина. Однако заболевание печени развивается лишь в случае мутации, при которой а,-антитрипсин накапливается в гепатоцитах. Классическим примером может служить вариант PiZZ, однако аналогичная картина возможна при Мmalton и Мduarte.
ПАТОГЕНЕЗ ПОРАЖЕНИЯ ПЕЧЕНИ [18]
Только фенотип PiZZ достоверно связан с заболеванием печени. Оно не обусловлено низкой активностью циркулирующего a1-антитрипсина, поступающего в печень, поскольку при других фенотипах с низкой активностью циркулирующего a1-антитрипсина повреждение печени не развивается. По-видимому, поражение печени обусловлено внутрипеченочным накоплением a1-антитрипсина. Исследования молекулярной структуры
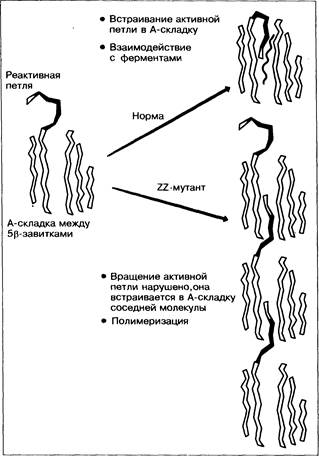
Рис. 23-16. Предполагаемый механизм полимеризации ZZ-a1-антитрипсина.
показали, что при ZZ-мутации происходит полимеризация белковых единиц. В норме активная петля (рис. 23-16) заходит между b-завитками так называемой А-складки белка и он взаимодействует с эластазой и другими ферментами. В случае мутантного ZZ-белка проникновение петли с активным центром невозможно. Она остаётся снаружи и поэтому приобретает способность включаться в А-складку соседней ZZ-молекулы [14]. Из-за полимеризации большая часть a1-антитрипсина остаётся в гепатоците.
Предполагают, что поражение печени обусловлено накоплением белка, однако механизм окончательно не выяснен. Полимеризация ZZ-белка происходит спонтанно или под влиянием провоцирующих факторов, например при повышении температуры. Однако мутация a1-антитрипсина — не единственная причина его накопления. В клетках, полученных от больного с недостаточностью а, -антитрипсина и поражением печени, было выявлено также угнетение разрушения ZZ-белка в эндоплазматической сети [18]. Таким образом, вариант клинического течения зависит не только от аномального белка, вырабатываемого у больных с PiZZ, но также и от других клеточных механизмов, пока ещё мало изученных. Трансгенные мыши с человеческим ZZ-типом белка — адекватный объект для изучения патогенеза этого заболевания [2].
Недостаточность a1-антитрипсина сопровождается широким спектром клинических проявлений. Число больных с распознанным печёночным или лёгочным поражением значительно меньше того, которое можно было бы ожидать, учитывая частоту встречаемости гена. Клинические проявления поражения печени разнообразны: от печёночной недостаточности и необходимости трансплантации в детском возрасте до исчезновения признаков заболевания печени к 18 годам — наиболее частого исхода заболевания [17]. Объяснением этому может служить влияние факторов окружающей среды и ещё неизвестных генетических факторов.
Наиболее частым проявлением недостаточности a1-антитрипсина является эмфизема, однако её клинические признаки наблюдаются только через десятки лет от начала заболевания [6, 17]. Существует пороговый уровень активности a1-антитрипсина, ниже которого риск заболевания увеличивается. Курение ускоряет развитие эмфиземы лёгких и уменьшает продолжительность жизни больных. Симптомы a1-антитрипсинзависимой эмфиземы лёгких обычно не появляются до третьего десятилетия жизни. Однако частота и тяжесть заболевания могут значительно варьировать; у некоторых курильщиков заболевание протекает бессимптомно либо симптомы его развиваются в седьмом или восьмом десятилетии жизни. Продолжительность жизни больных с недостаточностью а,-антитрипсина по сравнению с показателем в популяции снижена на 10—15 лет [6].
У большинства больных с вариантом PiZZ на определённом этапе жизни развивается заболевание печени. В первый год жизни активность АлАТ в сыворотке повышена у 75% детей [17]. В первые месяцы жизни у ряда больных развивается тяжёлая паренхиматозная желтуха, которая может привести к смерти или потребовать трансплантации печени. Однако большинство больных выздоравливают. Из 127 шведских детей, у которых при скрининговом обследовании выявлен PiZZ, у 22 заболевание печени клинически проявилось в грудном возрасте (неонатальный холестаз или гепатоспленомегалия). От цирроза в раннем возрасте погибли 2 больных; у 2 больных, умерших от других причин, при гистологическом исследовании выявлен цирроз или фиброз. Это согласуется с расчётными данными, согласно которым трансплантация печени требуется 3% детей с недостаточностью a1-антитрипсина [16]. Динамическое наблюдение за остальными детьми до 18-летнего возраста показало, что состояние их было удовлетворительным. Нарушение показателей функции печени было выявлено только у 2 из них [17].
Цирроз может оставаться компенсированным на протяжении многих лет, но может и неуклонно прогрессировать, приводя к смерти в детском возрасте в 25% случаев |1|. Частота поражения печени к 50-летнему возрасту составляет около 15%, среди мужчин она выше [4|. Первыми признаками заболевания могут быть портальная гипертензия или асцит.
В гепатологической клинике для взрослых среди 469 больных хроническими заболеваниями печени было выявлено только 5 гомозигот с недостаточностью a1-антитрипсина (фенотип ZZ), и у всех 5 в анамнезе была неонатальная желтуха [9|.
У одного и того же больного с недостаточностью a1-антитрипсина лёгкие и печень редко бывают поражены одновременно [10].
Заболевание, особенно у мужчин, может осложниться развитием ГЦК |8].
У больных с наследственным гемохроматозом чаще выявляют ген a1-антитрипсина, однако причина этой взаимосвязи не выяснена [7].
Гетерозиготы (MZ) чаще встречаются среди больных с криптогенным циррозом и хроническим гепатитом |3]. Значение этого факта неизвестно. У гетерозигот по недостаточности a1-антитрипсина, страдающих циррозом печени, возможно развитие ГЦК, однако скорее всего это связано с другими факторами, такими как вирусный гепатит С или алкоголизм, чем непосредственно с недостаточностью a1-антитрипсина [15].
Причиной поражения печени может быть также частичная недостаточность другого ингибитора протеаз — a1-антихимотрипсина [13].
ГИСТОЛОГИЧЕСКОЕ ИССЛЕДОВАНИЕ ПЕЧЕНИ
Заболевание при остром начале напоминает неонатальный гепатит, с тем лишь отличием, что не определяются гигантские клетки. Через 12 нед в перипортальных гепатоцитах обнаруживаются резистентные к действию диастазы и окрашивающиеся отчётливо в ШИК-реакции внутриклеточные глобулы (рис. 23-17), которые также специфически окрашиваются в реакции с a1-антитрипсиниммунопероксидазой. В печени повышено содержание меди.
При электронной микроскопии в расширенной шероховатой эндоплазматической сети видны скопления белка, флюоресцирующие при обработке антителами против a1-антитрипсина.
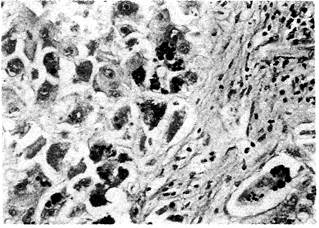
Рис. 23-17. Недостаточность a1-антитрипсина. В биоптате печени после обработки диастазой в ШИК-реакции выявляются ярко-красные депозиты в перипортальных гепатоцитах. См. также цветную иллюстрацию на с. 784.